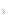 (Carregando Índice)... (Carregando Índice)... |
Última revisão: 15/01/2015
Comentários de assinantes: 0
Susan E. Haynes, MD,
Heather L. Bartlett, MD,
Larry T. Mahoney, MD,
David J. Skorton, MD
[The original English language work has been published by DECKER INTELLECTUAL PROPERTIES INC. Hamilton, Ontario, Canada. Copyright © 2014 Decker Intellectual Properties Inc. All Rights Reserved.]
Tradução: Soraya Imon de Oliveira.
Revisão técnica: Dr. Rodrigo Brandão Neto.
As doenças congênitas do coração e da vasculatura são os defeitos inatos mais comuns, ocorrendo em cerca de 8 a cada 1.000 bebês nascidos. Alguns pacientes com defeitos cardíacos congênitos (DCCs) usufruem, por muitos anos, de uma alta qualidade de vida; graças aos avanços médicos e cirúrgicos alcançados nas últimas décadas. Esta incidência, relativamente alta, acoplada a um tratamento aprimorado, resulta em um grande percentual de pacientes adultos com cardiopatias congenitas (CC). Atualmente, um número maior de adultos, comparado ao número de crianças, vivem com CC e este dado continuará crescendo.1 Hoje, uma vasta gama de clínicos tem de se manter bem informada acerca do cuidado para estes pacientes. Este capítulo, então, revisa as CCs mais encontradas em pacientes adultos.
Os defeitos de septo atrial (DSAs) ou comunicações intra-atriais ( CIAs) ocorrem em três localizações principais [ver Figura 1]: a região da fossa oval (DSAs de forame secundum); porção superior do septo atrial, próximo à junção com a veia cava superior (VCS) (DSAs de seio venoso); e porção inferior do septo atrial, próximo ao ânulo da valva tricúspide (DSAs de forame primum).2 Os DSAs de forame primum são considerados parte do espectro de defeitos septais atrioventriculares (DSAVs) [ver Defeitos Septais Atrioventriculares, adiante].
Os DSAs de forame secundum são a variedade mais comum e correspondem a mais da metade dos DSAs. Um defeito acompanhante frequente é o prolapso da valva mitral. O defeito de seio venoso é, relativamente, menos prevalente. O retorno venoso pulmonar anômalo é uma anomalia associada comum e a proximidade entre o nodo sinoatrial e o DSA pode acarretar disfunção do nodo sinoatrial e desenvolvimento de arritmias atriais.
Os DSAs ou CIAs estão associados a desvios da esquerda-direita de diversos graus;os principais determinantes da direção e magnitude do fluxo do desvio são: (i) o tamanho do defeito e (ii) as relativas complacências dos ventrículos esquerdo e direito.3
Alguns pacientes com DSAs tipo forame secundum, ou de seio venoso, permanecem assintomáticos durante a fase juvenil, ainda que a ecocardiografia antecipe o diagnóstico em alguns casos. O paciente ao se aproximar da meia-idade, passa a apresentar a complacência do ventrículo esquerdo diminuída, aumentando a magnitude do desvio da esquerda-direita. A dilatação atrial de longa duração pode levar a uma variedade de arritmias atriais, incluindo contrações atriais precoces, taquicardia supraventricular e fibrilação atrial. Um número substancial de pacientes de meia-idade relata dispneia, sobretudo ao esforço, até mesmo, na ausência de hipertensão pulmonar. Cerca de 10% dos pacientes com DSAs de forame secundum evoluem para hipertensão pulmonar associada à doença obstrutiva vascular pulmonar (síndrome de Eisenmenger) [ver Síndrome de Eisenmenger, adiante]. À medida que a pressão pulmonar sobe, o desvio da esquerda-direita irá diminuir e será substituído por um desvio direita-esquerda, resultando em cianose.
A principal característica observada ao exame físico de um indivíduo com DSA é uma ampla separação da segunda bulha cardíaca. É comum que haja um sopro sistólico a partir do fluxo pulmonar aumentado e, se um amplo desvio da esquerda-direita estiver presente, o fluxo adicional através da valva tricúspide pode levar um borborigmo diastólico reminiscente de estenose tricúspide.
Eletrocardiografia. O eixo QRS usualmente é normal no DSA tipo forame secundum, mas pode estar discretamente deslocado à direita, sendo que um padrão rSR’ é comum nas derivações precordiais direitas. No DSA tipo seio venoso, o eixo poderá estar normal ou relativamente horizontal (< 30°). Os ritmos atriais ectópicos ou outras evidências de disfunção do nodo sinoatrial poderão ser observadas.
Exames radiológicos. O raio x revela o alargamento do átrio direito, ventrículo direito e artéria pulmonar principal. Os vasos pulmonares exibem alargamento difuso, devido ao aumentado fluxo sanguíneo pulmonar. A imagem da ressonância magnética (IRM) com angiografia ou cateterismo cardíaco identificarão as veias pulmonares anômalas. Estas modalidades deverão ser consideradas quando houver suspeita da presença desta anomalia associada em pacientes com DSA de seio venoso.
O paciente com DSA tipo ostium secundum que tem hipertensão pulmonar poderá ser beneficiado pelo cateterismo cardíaco de lado direito, para determinação do nível de resistência e pressão arterial pulmonar.
Ecocardiografia. A ecocardiografia poderá confirmar a presença de um DSA, determinar seu tamanho e tipo, permitir o cálculo do fluxo de desvio através dele, e identificar quaisquer anomalias associadas.
Os DSAs amplos (definidos como sendo defeitos associados a uma proporção de fluxo pulmonar:sistêmico [Qp:Qs] > 1,5:1) deverão ser fechados para prevenir o desenvolvimento de hipertensão pulmonar e diminuir o risco de êmbolos paradoxais. O fechamento cirúrgico direto tem sido o método tradicionalmente empregado, porém, atualmente, existem dispositivos comumente disponibilizados que possibilitam o fechamento à base de cateterismo de muitos defeitos de forame secundum.4,5 O manejo pós-tratamento inclui a avaliação periódica para detecção de arritmias atriais.6 Os pacientes que passaram por correção do defeito transcateter requerem seguimento vitalício, devido ao risco de erosão cardíaca ou fratura da estrutura do dispositivo. Uma dor torácica súbita ou uma síncope inesperada deverão levar, o paciente, prontamente a avaliações adicionais com ecocardiografia, para examinaruma possível perfuração cardíaca.7

Figura 1 - Anatomia dos defeitos de septo atrial. DSAFP = defeito de septo atrial de forame primum; DSAFS = defeito de septo atrial de forma secundum; DSASV = defeito de septo atrial de seio venoso.
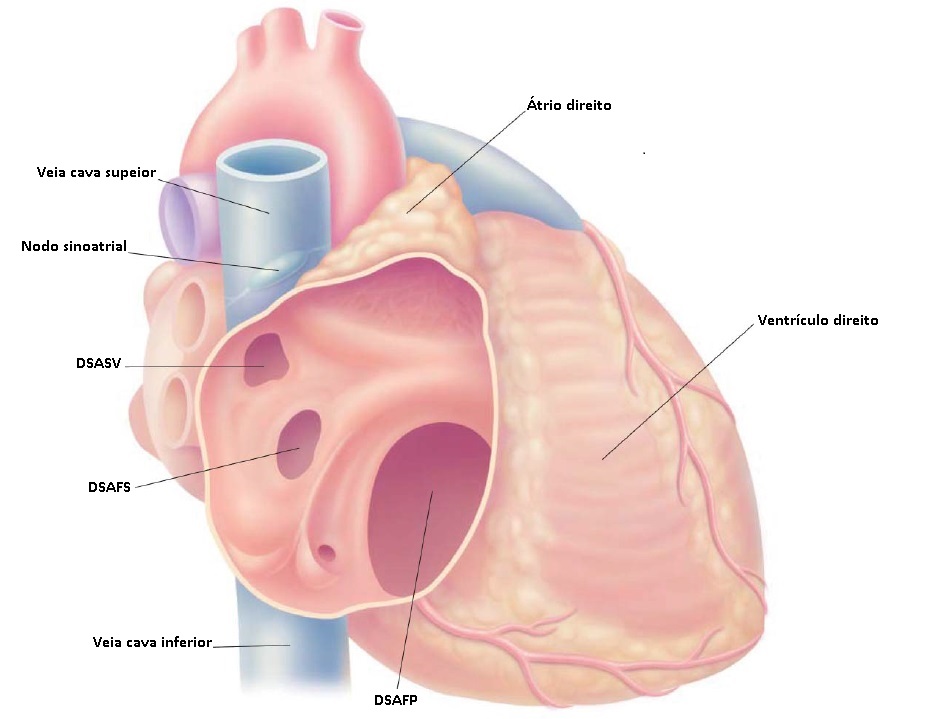
O folheto septal da valva tricúspide, normalmente, se insere no septo; em uma localização que é discretamente mais próxima do ápice, em comparação ao folheto septal da valva mitral [ver Figura 2]. Assim, a pequena porção do tecido septal superior à inserção do folheto septal tricúspide separa o átrio direito do ventrículo esquerdo, sendo, assim, denominada de septo atrioventricular. O termo DSAV refere-se a um complexo espectro de distúrbios envolvendo anormalidades do septo atrioventricular e, frequentemente, as valvas atrioventriculares. A nomenclatura para este espectro de distúrbios tem sido variável. Entre os termos sinônimos, estão o defeito de canal atrioventricular e o defeito do coxim endocárdico.
O espectro de DSAVs engloba desde um DSA primum simples, até um DSAV completo, permitindo a comunicação livre entre as quatro câmaras cardíacas. As variações anatômicas do folheto anterior da valva mitral e do folheto septal da valva tricúspide incluem uma fenda ou outra anomalia em qualquer um ou em ambos os folhetos; as cordas acessórias que se predem em localizações anômalas podem alterar a função dos folhetos valvulares ou produzir obstrução do trato de saída ventricular esquerdo; ou ainda um folheto valvar atrioventricular comum que comunica o defeito de septo. As consequências fisiológicas variam de acordo com a extensão da anomalia. Exemplificando-as, a adição de um folheto anterior de valva mitral fendido acrescenta graus variáveis de regurgitação mitral. Os defeitos maiores que, também, envolvem o septo ventricular, bem como os DSAVs completos poderão estar associados aos desvios da esquerda-direita torrenciais ou a uma mistura de sangue venoso e arterial.
Pacientes com DSAV completo não reparado apresentam risco de desenvolvimento de hipertensão pulmonar. A síndrome de Eisenmenger é, particularmente, comum em pacientes com DSAV e com síndrome de Down (trissomia do 21).8
Figura 2 - Corte transversal anatômico mostrando o septo atrioventricular (SAV). Note que o folheto septal da valva tricúspide está inserido mais proximamente do ápice, em comparação ao ponto de inserção do folheto septal da valva mitral. Por este motivo, o SAV separa o átrio direito do ventrículo esquerdo.
Pacientes com DSAs tipo forame primum isolados podem ser assintomáticos até atingirem a idade adulta, quando, então, poderão apresentar fadiga, dispneia ou sintomas relacionados a arritmias atriais. A intensa regurgitação valvar atrioventricular poderá produzir sintomas de insuficiência cardíaca ou arritmias. Os sintomas relacionados à hipertensão pulmonar são observados em pacientes que desenvolvem a síndrome de Eisenmenger.
Os pacientes que apresentam apenas um DSA de forame primum apresentam achados clínicos similares àqueles encontrados em pacientes com DSA de forame secundum. Um sopro pansistólico adicional poderá ser encontrado em pacientes com insuficiência de valva atrioventricular ou defeito septal ventricular (DSV).
Eletrocardiografia. O desvio de eixo à esquerda está presente na maioria dos pacientes, a combinação dos achados físicos de DSA como desvio de eixo à esquerda ao eletrocardiograma (ECG) sugere a presença de um DSAV. O retardo de condução ventricular direita e a hipertrofia ventricular, também ,poderão estar presentes.
Exames radiológicos. O raio-x mostra cardiomegalia e ingurgitamento vascular pulmonar, em decorrência do desvio da esquerda-direita.
Ecocardiografia. A ecocardiografia define, especificamente, a anatomia e a importância funcional dos defeitos. A avaliação ecocardiográfica pré-operatória inclui uma estimativa da gravidade da regurgitação valvar atrioventricular, a proporção Qp:Qs e as pressões arteriais pulmonares. No pós-operatório, a ecocardiografia é usada para identificar e avaliar a importância da estenose/regurgitação valvar atrioventricular ou desvio residual.
O raro paciente que apresenta DSAV completo deve ser avaliado para hipertensão pulmonar. Se a presença de hipertensão pulmonar não for proibitiva (i. e., resistência vascular pulmonar < 50% da resistência vascular sistêmica), então o fechamento cirúrgico do defeito e o reparo das anomalias da valva atrioventricular deverão ser realizados. No pós-operatório, os pacientes são avaliados quanto à adequação do reparo da valva atrioventricular e monitorados para detecção de evidências de desvio residual. Em pacientes com regurgitação mitral residual, o tratamento enfoca a terapia médica versus a necessidade e o momento para realização de uma nova operação, que poderá envolver o reparo ou a substituição da valva mitral. O paciente, também, deverá receber seguimento para desenvolvimento de arritmias atriais.
Os DSVs estão entre os distúrbios cardíacos congênitos mais encontrados ao nascimento, embora sejam vistos com menor frequência como lesões isoladas durante a fase adulta. Isto ocorre , pois a maioria dos DSVs em bebês é (1) ampla ou não restritiva (i.e., permitem o equilíbrio de pressões entre os ventrículos) e, com isso levando o paciente à insuficiência cardíaca, necessitando, assim, de um fechamento cirúrgico antecipado, ou (2) pequena e se fecha espontaneamente.
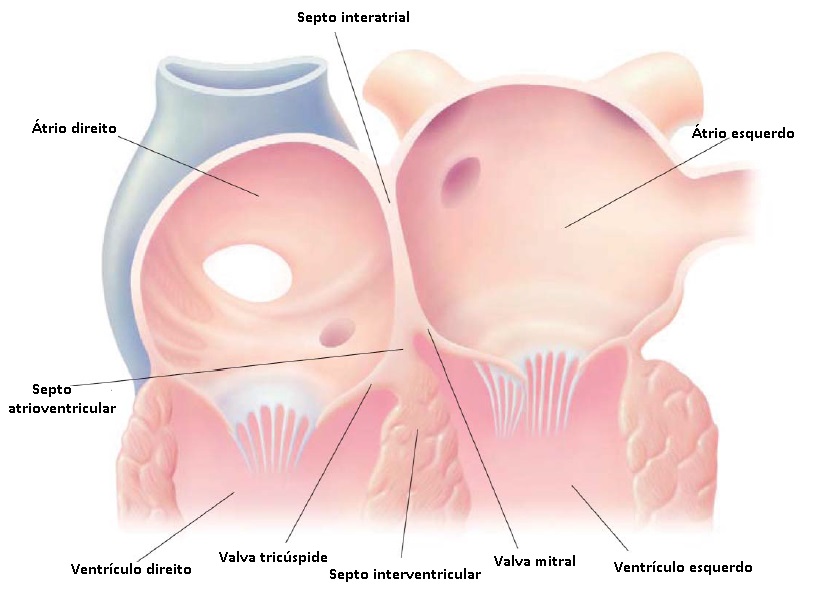
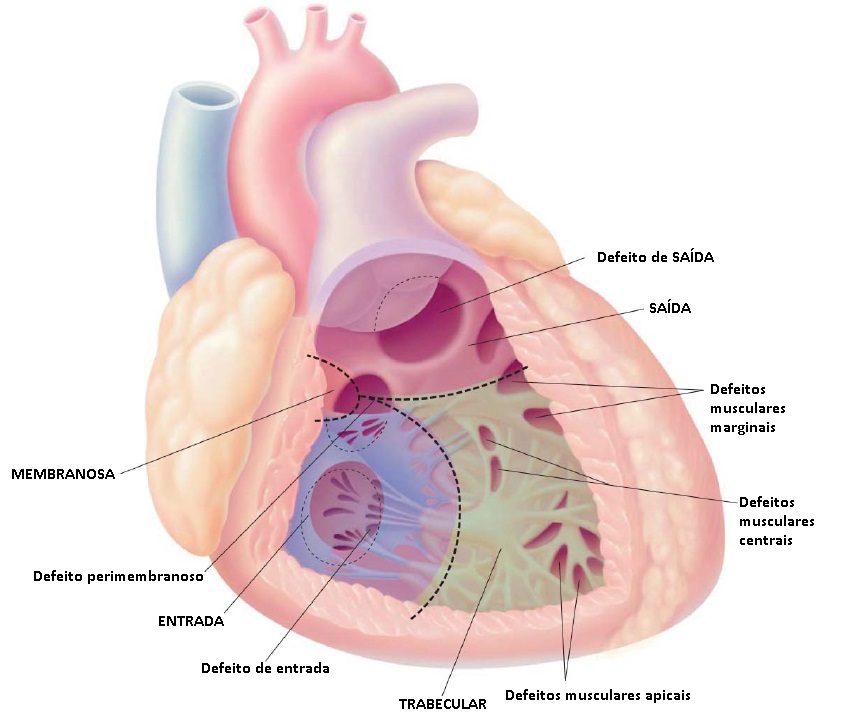
Os sistemas de classificação para DSV variam, mas usualmente fazem referência às divisões embriológicas do septo ventricular nas porções de entrada, saída, muscular e membranosa [ver Figura 3]. Os defeitos mais comuns são os perimembranosos. Os DSVs de entrada, de localização mais posterior, podem fazer parte do espectro de DSAVs (ver anteriormente). Defeitos únicos ou múltiplos ocorrerem no septo muscular (DSV muscular). Por fim, os DSVs de saída incluem os defeitos subpulmonares, que poderão propiciar o prolapso de uma cúspide aórtica, levando à associada regurgitação aórtica.
Os DSVs não restritivos permitem o equilíbrio de pressões entre os ventrículos direito e esquerdo, enquanto os defeitos pequenos produzem um amplo gradiente de pressão ao longo do defeito, de tal modo que as pressões no coração direito permanecem normais. A magnitude do fluxo de desvio através de DSVs moderados ou amplos depende das resistências relativas do leito vascular sistêmico versus pulmonar. Em casos raros, o médico poderá se deparar com pacientes adultos que possuem defeitos amplos e não restritivos na ausência de outras lesões. A estenose pulmônica moderada ao nível valvar ou subvalvar poderá criar uma resistência aumentada ao fluxo de saída ventricular direito suficiente para diminuir o desvio da esquerda-direita. Em consequência, os pacientes com DSV e estenose pulmônica leve a moderada poderão atingir a fase adulta sem uma manifestação sintomática. Os adultos com DSV de longa duração e desvios amplos poderão desenvolver síndrome de Eisenmenger.
Com exceção dos pacientes que contraem endocardite infecciosa ou daqueles com síndrome de Eisenmenger, os adultos com DSV são assintomáticos.
O clássico achado físico de um DSV restritivo é um sopro pansistólico áspero, frequentemente palpável, que é melhor ouvido na borda external inferior esquerda. Os pacientes com defeitos amplos que possibilitam o equilíbrio das pressões ventriculares poderão apresentar sopros menos impressionantes do que aqueles encontrados em pacientes com defeitos pequenos. O motivo para isto é que, com os defeitos pequenos, existe um amplo gradiente entre os ventrículos esquerdo e direito, que resulta numa forte turbulência ao longo do defeito. Quando há prolapso da cúspide aórtica, o sopro da regurgitação aórtica é audível.
Eletrocardiografia. O ECG poderá ser normal ou mostrar evidência de ampliação atrial esquerda ou de hipertrofia ventricular esquerda (HVE), além de um padrão da chamada sobrecarga diastólica, caracterizado por ondas Q proeminentes nas derivações precordiais da esquerda (V5 e V6) e nas derivações I e aVL. Após o fechamento cirúrgico de um DSV, é possível observar um bloqueio de feixe direito.
Exames radiológicos. O raio-x poderá estar normal ou poderá mostrar um alargamento ventricular esquerdo e um ingurgitamento arterial pulmonar. Os pacientes que apresentam evidência de hipertensão pulmonar deverão ser submetidos ao cateterismo cardíaco de lado direito, a fim de determinar o grau de hipertensão pulmonar e o nível de qualquer obstrução pulmônica.
Ecocardiografia. A ecocardiografia é o exame de escolha para identificar a localização, tamanho e relevância hemodinâmica de um DSV ou quaisquer defeitos associados. O gradiente interventricular deverá ser determinado para estimar a pressão ventricular direita. Além disso, será preciso realizar uma avaliação do fluxo sanguíneo pulmonar aumentado.
Os pacientes com DSVs com uma proporção Qp:Qs > 1.5:1 deverão ser considerados candidatos ao fechamento cirúrgico. Os pacientes com hipertensão pulmonar poderão ser submetidos ao fechamento se a resistência pulmonar não exceder 50% da resistência sistêmica. O prolapso da cúspide aórtica com consequente regurgitação aórtica pode diminuir a magnitude do desvio, contudo a presença de um prolapso constitui uma potencial indicação adicional para o fechamento.
Os fechamentos operatórios antecipados de DSV eram realizados por ventriculotomia direita. Hoje, porém, a maioria dos defeitos (em particular os defeitos no septo perimembranoso) é fechada através de uma abordagem transatrial. Este tipo de abordagem gera menos problemas de arritmias e disfunção ventricular direita. Avanços contínuos estão sendo alcançados em termos de desenvolvimento de dispositivos de fechamento transcateter. Atualmente, porém, o fechamento cirúrgico do DSV continua sendo a abordagem mais comum. O tratamento pós-fechamento envolve avaliação para DSV residual ou recorrente e arritmias atriais ou ventriculares, bem como avaliação da função ventricular direita.
Durante a vida fetal, o canal arterial conecta a artéria pulmonar à aorta. Logo após o nascimento, como resultado das alterações dos níveis circulantes de prostaglandina e da saturação do oxigênio arterial, esse canal sofre constrição seguida de fechamento permanente. A falha do fechamento do canal arterial leva ao desenvolvimento de uma condição denominada canal arterial persistente (CAP).
Figura 3 - Posições anatômicas de defeitos septais ventriculares (DSVs). As principais subdivisões anatômicas do septo ventricular são as porções membranosa, saída, entrada e trabecular. DSVs típicos: defeito de saída, defeito perimembranoso, defeitos musculares marginais, defeitos musculares centrais, defeito de entrada e defeitos musculares apicais.
O desvio da aorta para a artéria pulmonar aumenta o fluxo sanguíneo pulmonar e o retorno para o lado esquerdo do coração. O tamanho do defeito e as resistências relativas dos leitos vasculares sistêmico e pulmonar determinam o grau de desvio. Os adultos que têm CAP ,comumente, apresentam uma lesão pequena sem desvio amplo da esquerda-direita ou exibem lesões maiores e síndrome de Eisenmenger.
Com exceção dos pacientes com síndrome de Eisenmenger, a maioria dos adultos com CAP pequeno a moderado é assintomática, a menos que haja desenvolvimento de endarterite.
O achado físico patognomônico de CAP é o sopro contínuo; um sopro contínuo é aquele que é audível ao longo de toda a sístole e diástole adentro, em qualquer extensão. O clássico sopro de CAP é robótico e se estende por toda a sístole até graus variáveis na diástole, atingindo o pico de intensidade no momento de S2. A saída do sangue para dentro da artéria pulmonar na diástole produzirá uma ampla pressão de pulsação, devido à baixa pressão diastólica aórtica.
Eletrocardiografia. O ECG em pacientes com CAP pode ser normal ou mostrar evidência de HVE.
Exames radiológicos. Se o desvio for pequeno, o raio-x torácico pode ser normal. Os pacientes com desvios maiores terão cardiomegalia associada e marcas vasculares aumentadas. Em adultos, é possível notar a presença de cálcio junto á parede do canal.
Ecocardiografia. A ecocardiografia identificará o CAP e permitirá a quantificação da proporção Qp:Qs.
Com o advento das técnicas confiáveis de fechamento transcateter de CAPs, a prática comum é recomendar que a maioria dos CAPs seja fechada.9 Em casos raros, o canal precisará ser fechado cirurgicamente; caso o fechamento transcateter fracasse. O tratamento pós-operatório inclui a avaliação de um desvio residual, embora isto seja incomum. Os pacientes que desenvolvem hipertensão pulmonar são tratados do mesmo modo que aqueles com síndrome de Eisenmenger [ver síndrome de Eisenmenger, adiante].
As anormalidades do trato de saída ventricular esquerdo são distúrbios cardíacos congênitos comuns. Em particular, até 2% da população tem valvas aórticas bicúspides congênitas. Uma valva aórtica bicúspide pode ser descoberta como um achado acidental ao exame físico ou na ecocardiografia realizada por outros motivos, mas pode se tornar estenosada ou regurgitante com o passar do tempo. As valvas aórticas bicúspides poderão estar associadas à coarctação da aorta.
Algumas valvas aórticas bicúspides não se tornam estenóticas nem regurgitantes. Entretanto, uma valva aórtica bicúspide estenosada produzirá uma sobrecarga de pressão ventricular esquerda, que resultará em HVE e, se grave, poderá levar eventualmente à insuficiência cardíaca, angina de peito ou morte súbita por taquiarritmias. O paciente com uma valva aórtica bicúspide incompetente exibirá dilatação do ventrículo esquerdo, a princípio com função sistólica normal. A condição posteriormente poderá evoluir para insuficiência cardíaca. Mesmo na ausência de obstrução significativa, a dilatação da aorta ascendente é observada com frequência associada a uma valva aórtica bicúspide.10
Ao exame físico, o sinal cardinal de uma valva aórtica bicúspide é um clique de ejeção sistólica precoce. A qualidade e duração dos sopros, frequentemente, estão correlacionadas com a gravidade da estenose ou regurgitação aórticas. Na ausência de uma anomalia hemodinâmica significativa, nenhum sopro ou apenas um sopro de ejeção suave é audível. Um sopro bastante discreto de regurgitação aórtica não é incomum, mesmo com valvas aórticas bicúspides hemodinamicamente irrelevantes.
Eletrocardiografia. O ECG será normal, a menos que uma estenose ou regurgitação hemodinamicamente significativa esteja presente. Neste caso, o ECG mostrará HVE.
Exames radiológicos. Os achados do raio-x de torax em pacientes com valvas aórticas bicúspides hemodinamicamente significativas são similares àqueles encontrados em pacientes com estenose aórtica e regurgitação aórtica de outras etiologias. Estas podem incluir a dilatação do ventrículo esquerdo na regurgitação aórtica ou na estenose aórtica que esteja progredindo para insuficiência cardíaca; esta última condição, também, produzirá congestão pulmonar.
Ecocardiografia. Tanto a presença de uma valva aórtica bicúspide como a sua relevância hemodinâmica poderão ser determinadas pela ecocardiografia. Exames seriados serão úteis no acompanhamento da evolução da lesão e da função do ventrículo esquerdo. A dimensão da aorta ascendente pode ser medida.11
Os pacientes com regurgitação aórtica a partir de uma valva bicúspide que sejam assintomáticos e apresentem função sistólica normal são seguidos com anamnese, exames físicos e ecocardiogramas a intervalos regulares. Se a qualidade precária das imagens acústicas comprometer a confiabilidade das determinações ecocardiográficas, a IRM poderá ser considerada. Havendo sintomas de insuficiência cardíaca, evidência de diminuição da função sistólica ou dilatação progressiva do ventrículo esquerdo, a substituição cirúrgica da valva aórtica é indicada.
A valvuloplastia cirúrgica ou por balão (em pacientes mais jovens) deve ser considerada em casos de estenose aórtica grave, especialmente quando acompanhada de insuficiência cardíaca, síncope ou desconforto torácico. Existem diversos procedimentos cirúrgicos disponíveis, incluindo o reparo direto da valva, substituição com bioprótese ou prótese mecânica, substituição da valva e raiz aórtica proximal, e procedimento de Ross (remoção cirúrgica da valva bicúspide anormal seguida de substituição pela valva pulmônica nativa do próprio paciente que, por sua vez, é substituída por um tecido valvar). O procedimento de Ross elimina a necessidade de uma valva artificial na posição aórtica. O tratamento pós-operatório enfoca a avaliação da estenose recorrente ou regurgitação aórtica progressiva.
Os pacientes com estenose pulmonar comumente apresentam uma valva deformada, em que a fusão de uma ou mais comissuras resulta em uma valva em forma de cúpula. A maioria destas valvas é delgada e flexível. Entretanto, alguns pacientes apresentam valvas espessas , denominadas displásica
A valva pulmonar estenótica impõe uma carga de pressão ao ventrículo direito, levando à hipertrofia ventricular direita (HVD) e, em um subgrupo de pacientes, insuficiência ventricular direita. Em pacientes com ventrículo direito gravemente hipertrófico, o desequilíbrio entre suprimento e demanda de oxigênio miocárdico poderá levar à isquemia, acompanhada de desconforto torácico anginal e arritmias.
Os pacientes com estenose pulmonar, ainda que de grau moderadamente sério, poderão permanecerem assintomáticos por décadas. Os sintomas eventuais incluem um desconforto torácico reminiscente de angina de peito decorrente de arteriopatia coronariana; falta de ar; fadigabilidade e sintomas de insuficiência ventricular direita. Após a adolescência, a evolução da doença e manifestação dos sintomas são incomuns.
O achado físico cardinal de estenose pulmonar é um sopro sistólico em crescente-decrescente de turbulência através da valva estreitada, que é precedido por um clique de ejeção pulmônica. O comportamento do clique de ejeção pulmonar durante a respiração pode ser útil para diferenciá-lo do clique da valva aórtica bicúspide. O clique de ejeção exibirá uma diminuição seletiva de intensidade com inspiração normal, podendo, até, desaparecer totalmente com a inspiração. Em contraste, o clique da valva aórtica bicúspide não exibirá essa diminuição seletiva.
Eletrocardiografia. O ECG será normal em pacientes com estenose pulmonar leve a moderada. A estenose grave conduz à HVD.
Exames radiológicos. Em pacientes com graus leves a moderados de estenose pulmonar, é possível que o raio-x torácico somente mostre uma discreta dilatação pós-estenótica do tronco pulmonar proximal.
Ecocardiografia. A ecocardiografia é extremamente precisa para identificar e diagnosticar a gravidade da estenose pulmonar, além de, também, conseguir diferenciar entre valvas pulmonares flexíveis e displásicas. Por fim, o ecocardiograma poderá ser usado para avaliar semiqualitativamente a função sistólica ventricular direita.
As primeiras abordagens para estenose pulmonar consistiam em uma valvotomia cirúrgica fechada ou aberta. Nos últimos 20 anos, o advento dos métodos confiáveis de valvuloplastia pulmonar com balão propiciaram uma mudança significativa na abordagem destes casos.4,12 A primeira linha de tratamento para a estenose de valva pulmonar consiste na realização de uma valvuloplastia com balão, em casos de estenose significativa (definida por um gradiente de trato de saída ventricular direito superior a 50 mmHg). Algumas valvas displásicas são difíceis de tratar adequadamente por valvuloplastia com balão e requerem reparo ou substituição cirúrgica. O tratamento pós-operatório inclui vigília contra estenose recorrente ou regurgitação progressiva.13 Os pacientes eventualmente pode requerer substituição cirúrgica da valva.
A coarctação da aorta é uma CC relativamente comum que pode ser isolada, acompanhada de outros defeitos (em especial DSV) e constitui um achado cardíaco comum em pacientes com síndrome de Turner. A valva aórtica bicúspide é uma lesão associada comum. A coarctação é causa comum de hipertensão secundária e deverá ser investigada em todos os pacientes que apresentam hipertensão sistêmica.
A patologia essencial, na coarctação da aorta, é o estreitamento do lúmen aórtico, usualmente nas adjacências do ligamento arterioso, distal ao ponto de partida da artéria subclávia esquerda. O estreitamento da aorta no sítio de coarctação divide a circulação sistêmica em uma zona de pressão alta proximal à coarctação e em uma zona de baixa pressão distal. A hipertensão poderá acelerar o desenvolvimento da arteriopatia coronariana aterosclerótica e levar a um acidente vascular encefálico. O acidente vascular encefálico representa, particularmente, um risco quando o indivíduo tem aneurismas do círculo de Willis, como ocorre na incidência aumentada de pacientes com coarctação.
Embora possa haver claudicação de membro inferior, os pacientes poderão permanecer totalmente assintomáticos, mesmo que haja uma significativa coarctação da aorta. Ao exame físico, o achado cardinal é a diferença de pulsos e pressões arteriais acima versus abaixo da coarctação. A palpação das artérias radial e femoral em um paciente normal revelará a chegada simultânea ou, talvez, discretamente antecipada do pulso na artéria femoral. Na coarctação da aorta, o pulso femoral será mais tardio do que na artéria radial e frequentemente com uma amplitude menor. A pressão arterial deve ser avaliada em ambos os braços e em qualquer perna durante a investigação da coarctação da aorta, devido às variações anatômicas. Quando a coarctação é distal a origem da artéria subclávia esquerda, ambos os braços estão na zona de alta pressão e ambas as pernas estarão na zona de baixa pressão. Entretanto, algumas coarctações são proximais à artéria subclávia esquerda. O braço esquerdo e ambas as pernas, portanto, estarão na zona de baixa pressão e o diagnóstico será perdido se apenas o braço esquerdo for usado para medir a pressão arterial. Mais raramente, a artéria subclávia direita pode ter uma origem anômala. Esta artéria, então, poderá surgir diretamente da aorta distal à artéria subclávia esquerda, e não da artéria braquiocefálica (inominada). Além das pressões arteriais diferenciais, um exame físico, também, poderá revelar um sopro ao longo da coarctação, mais bem ouvido na área infraescapular esquerda.
Eletrocardiografia. Em pacientes com coarctação, o ECG mostrará diversos graus de HVE, dependendo da gravidade do estreitamento.
Exames radiológicos. A dilatação da aorta proximal e distal ao sítio de coarctação poderá levar ao conhecido “sinal da árvore”, ao exame de raio x de tórax . Os adultos podem apresentar chanfradura de costela. Este termo refere-se à evidente obliteração – ou o chamado recorte – das bordas inferiores das costelas (usualmente, a 3ª a 9ª costela), devido aos amplos vasos colaterais intercostais de fluxo intenso que se desenvolvem como um mecanismo compensatório para desviar do estreitamento no sítio de coarctação. A ausência de chanfradura de costela, porém, não exclui a hipótese de coarctação da aorta.
A IRM cardíaca com angiografia poderá ser usada efetivamente para identificar a coarctação e a circulação colateral. Também, é útil na detectação de aneurismas ou de re-estenose no sítio de reparo. Os fatores de risco para as anomalias associadas de aorta ascendente e descendente incluem a valva aórtica bicúspide e a idade acima de 30 anos.14
Ecocardiografia. A ecocardiografia é extremamente útil para identificar o sítio de coarctação por visualização direta, bem como para medir o gradiente de pressão ao longo do sítio de coarctação. A ecocardiografia, também, consegue identificar uma valva aórtica bicúspide, que frequentemente acompanha a coarctação da aorta.
Uma coarctação que seja suficiente para produzir hipertensão deverá ser sempre tratada, seja por cirurgia ou por angioplastia com balão ou stent. Existe uma experiência mais vasta com o emprego da excisão cirúrgica da coarctação e com a anastomose de ponta-a-ponta ou interposição de enxerto. Nos últimos anos, tem sido crescentemente demonstrado que a angioplastia com balão e stent é uma alternativa viável para o tratamento inicial da coarctação e para tratamento da re-estenose no sítio de coarctação, que se desenvolve após o reparo ou a angioplastia.4 A angioplastia com stent de uma coarctação nativa ou recorrente pode resultar em menos complicações do que a cirurgia ou angioplastia com balão.15
A veia cava superior esquerda (VCSE) é uma estrutura embrionária persistente que não regride, normalmente, durante o desenvolvimento fetal. Isto ocorre em até 10% dos indivíduos com CC e poderá ocorrer como uma anomalia isolada em 0,3% da população.16
Durante o desenvolvimento embrionário, a veia cava anterior esquerda sofre obliteração e forma o ligamento de Marshall. Quando isto não ocorre, uma VCSE persistente drena para dentro do seio coronário [ver Figura 4]. Comumente, há, também, uma veia cava superior direita.17 O paciente não apresenta riscos nem sintomas. Entretanto, é possível que surjam dificuldades durante a colocação de cateteres centrais de inserção periférica a partir do membro superior esquerdo ou durante a implantação de um sistema marca-passo transvenoso. Na presença de um seio coronário sem teto, uma VCSE persistente drena efetivamente para dentro do átrio esquerdo, resultando em aumento do risco de embolia sistêmica por trombos venosos.
Exceto nos casos de uso de aparelhos, os pacientes tendem a ser completamente assintomáticos e a não apresentarem achados físicos sugestivos de VCSE. Os indivíduos com seio coronário sem teto (um desvio da esquerda-direita) podem apresentar graus variáveis de dessaturação, que muitas vezes são detectados de modo incidental.
Eletrocardiografia. Poderá haver um baixo ritmo atrial direito ou o ECG poderá ser normal (na ausência de CC associada).
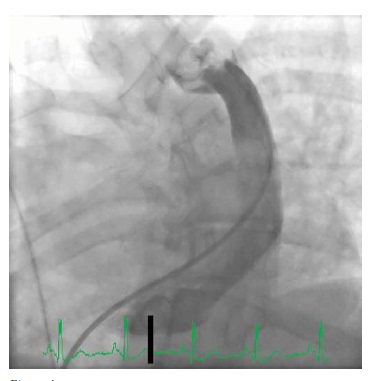
Figura 4 - Angiograma de uma veia cava superior esquerda persistente drenando para dentro do átrio direito.
Ecocardiografia. O ecocardiograma detectará a VCSE e demonstrará um seio coronário dilatado que recebe fluxo aumentado da VCSE.
Em geral, a VCSE persistente com um seio coronário intacto dispensa tratamento. A presença de um seio coronário defeituosos é indicação para reparo cirúrgico. Se houver uma veia de conexão, a VCSE poderá ser ligada sem que o seio coronário seja abordado. Na ausência de uma veia de conexão, o seio coronário tem de ser contido dentro do átrio direito.16
A cianose central é causada por um desvio da direita-esquerda intracardíaco ou intrapulmonar. A cianose se torna evidente quando os reduzidos níveis capilares de hemoglobina (não oxigenada) atingem cerca de 5 g/dL, embora isto dependa da concentração total de hemoglobina: a cianose é mais prontamente evidente em pacientes com policitemia e é menos evidente em pacientes com anemia. A cianose somente é evidenciada quando a saturação de oxigênio cai para menos de 85% (considerando níveis normais de hemoglobina). Os pacientes com dessaturação arterial de longa duração desenvolverão baqueteamento das unhas nos dedos das mãos e dos pés. O baqueteamento é caracterizado pelo espessamento e ampliação dos leitos ungueais, bem como pela perda do ângulo entre a unha e o leito ungueal que produz uma unha convexa. A escoliose, também,é comumente observada associada à cardiopatia cianótica.
É útil classificar as CCs cianóticas quanto aos efeitos que elas exercem sobre o fluxo sanguíneo pulmonar. Os defeitos produtores de diminuição do fluxo sanguíneo pulmonar incluem a tetralogia de Fallot, atresia tricúspide, anomalia de Ebstein e atresia pulmonar. Os defeitos associados ao aumento do fluxo sanguíneo pulmonar incluem o tronco arterial persistente, a transposição dos grandes vasos da base com ou sem DSV ou CAP, o retorno venoso pulmonar anômalo total, um ventrículo único ou comum, e a síndrome do coração esquerdo hipoplásico. Os pacientes acianóticos com amplos desvios da esquerda-direita podem desenvolver uma doença obstrutiva vascular pulmonar (síndrome de Eisenmenger).
Os pacientes adultos com CC cianótica apresentam risco aumentado de hiperviscosidade secundária à eritrocitose. A eritrocitose se desenvolve como um mecanismo compensatório para a dessaturação de oxigênio das hemácias: uma massa eritrocitária significativamente aumentada se faz necessária para que um volume adequado de oxigênio seja distribuído aos tecidos periféricos. A trombose venosa e arterial com acidentes vasculares encefálicos pode ocorrer na CC cianótica e foi atribuída à aumentada massa eritrocitária e anemia ferropriva associada, que também aumentam a viscosidade do sangue. Este risco é aumentado na presença de hipertensão ou fibrilação atrial, bem como em pacientes com história de flebotomia e microcitose, sugerindo a necessidade de uma abordagem mais conservativa para a flebotomia e de um tratamento agressivo da deficiência de ferro.
A tetralogia de Fallot é a forma mais comum de CC cianótica. Classicamente, a síndrome inclui estenose pulmonar (subvalvar, valvar, supravalvar ou uma combinação das três), HVD, DSV subaórtico e destroposicionamento da aorta, de tal modo que destrói o septo interventricular. As anomalias associadas incluem um arco aórtico direito (25%), DSA (10%) e anomalias arteriais coronarianas (10%).18 Cerca de 15% dos pacientes com tetralogia de Fallot apresentam deleção do cromossomo 22q11 (síndrome CATCH 22: anomalias cardíacas, fáscias anormais, hipoplasia tímica, fenda [cleft] palatina, hipocalcemia e deleção de 22q11).19
A prática cirúrgica vigente garante o reparo precoce, usualmente ainda no primeiro ano de vida;sem cirurgia, a sobrevida além de 20 anos de idade é incomum.
O reparo cirúrgico consiste no remendo de fechamento do DSV e alívio da obstrução do trato de saída ventricular direito com a adoção de um ou mais dos seguintes métodos: (i) ressecção do músculo infundibular; (ii) valvotomia pulmonar; (iii) ampliação do reparo transanular ou do trato de saída (iv) e ampliação do reparo do ramo principal ou proximal das artérias pulmonares. Em alguns casos, haverá a necessidade de colocação de um canal condutor desde o ventrículo direito até a artéria pulmonar. Esse canal poderá ter ou não valva, ser bioprotético ou um homoenxerto.
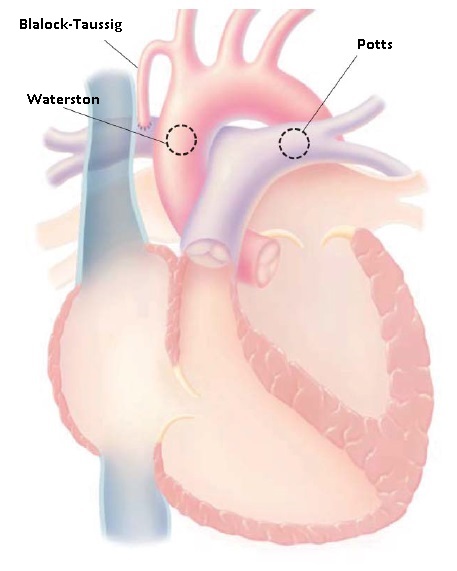
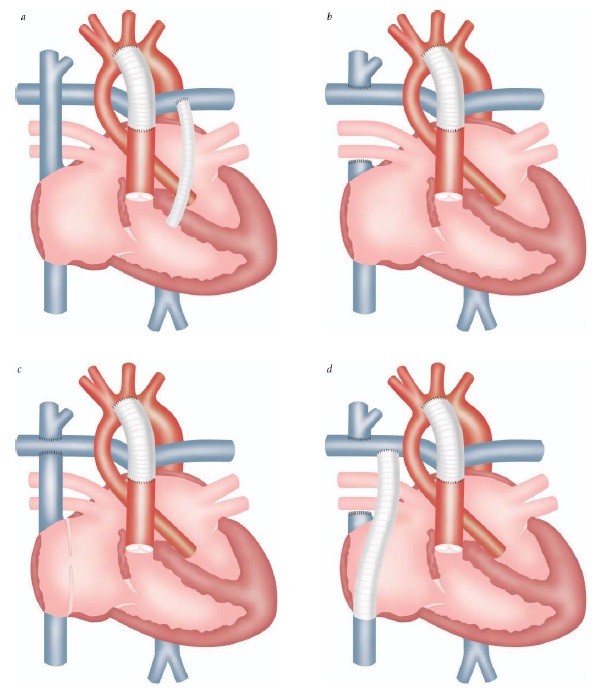
Embora o reparo precoce, nos dias de hoje, seja rotineiro, muitos pacientes são submetidos a procedimentos paliativos. A paliação cirúrgica poderá incluir um desvio da circulação sistêmica para a artéria pulmonar, com a meta de fornecer um fluxo pulmonar adicional. Este desvio pode ser um desvio de Blalock-Taussig , desvio de Potts ou desvio de Waterston. O clássico desvio de Blalock-Taussig conecta a artéria subclávia à artéria pulmonar, e sua forma modificada envolve a interposição de um enxerto de tubo, usualmente de politetra?uoroetileno estendido [Gore-Tex]. Um desvio de Potts conecta a aorta descendente à artéria pulmonar esquerda. Um desvio de Waterston conecta a aorta ascendente à artéria pulmonar direita [ver Figura 5].
Em pacientes submetidos ao reparo cirúrgico da tetralogia de Fallot, o exame enfoca os defeitos residuais. De forma não incomum, estes pacientes apresentam sopros relacionados à obstrução residual do trato de saída e uma regurgitação pulmonar leve à grave, que produz um sopro do tipo vaivém. Os sopros de ejeção sistólica poderão ser proeminentes, e estão mais relacionados com a técnica de reparo usada do que com o grau de obstrução. Os desvios permanentes produzirão um sopro contínuo. O grau de cianose dependerá da adequação do fluxo sanguíneo pulmonar proporcionada pelo desvio. Um DSA, DSV ou estenose de ramo arterial pulmonar residuais podem ser detectados. Com a crescente sobrecarga de volume ventricular direito a partir da obstrução ou insuficiência do fluxo de saída, o paciente pode apresentar intolerância ao exercício, insuficiência cardíaca direita, e arritmias atriais ou ventriculares.
Eletrocardiografia. Em pacientes submetidos ao reparo cirúrgico da tetralogia de Fallot, o ECG tipicamente mostra ritmo sinusal, desvio de eixo à direita e HVD. A maioria destes pacientes também apresenta bloqueio de feixe direito. Podem ser detectadas arritmias atriais e ventriculares, em especial em um exame de monitoramento 24 horas.
Exames radiológicos. Os achados obtidos com um raio x torácico variam de acordo com a história cirúrgica. Um arco aórtico de lado direito poderá ser notado. O segmento arterial pulmonar é côncavo, devido ao grau variável de hipoplasia da artéria pulmonar, enquanto a HVD resulta em um ápice virado para cima. Juntos, estes achados produzem o clássico achado de “coração em forma de bota”. A intervenção cirúrgica poderá resultar em regurgitação pulmonar significativa, que eventualmente levará a uma sobrecarga de volume cardíaco e consequente cardiomegalia. Com o passar do tempo, a ampliação do remendo do trato de saída pode se tornar aneurismática, conforme indicado por um segmento arterial pulmonar aumentado. Os canais condutores que seguem do ventrículo direito até a artéria pulmonar poderão apresentar calcificações. Um fluxo sanguíneo pulmonar assimétrico sugere a obstrução significativa do ramo arterial pulmonar, e a melhor forma de mensurá-lo é com uma avaliação do fluxo pulmonar. A IRM, padrão ouro dos exames de imagem, será útil na identificação dos defeitos residuais e da quantificaçãoda função ventricular direita, especialmente em pacientes com janelas acústicas precárias e exames ecocardiográficos inadequados [ver Figura 6]. Os critérios sugeridos para o momento adequado da substituição da valva pulmonar são baseados em várias medidas de IRM [ver Tabela 1].20
Ecocardiografia. A ecocardiografia estabelecerá a presença e gravidade de quaisquer defeitos residuais, incluindo a ampliação progressiva do ventrículo direito secundária à regurgitação pulmônica, um DSV residual e o fluxo contínuo em um desvio paliativo. Os exames de Doppler demonstrarão a magnitude do gradiente do trato de saída residual. A aorta ascendente, muitas vezes, está ampliada ao nascimento e continua sendo mais ampla por toda a vida. No entanto, as dimensões geralmente não atendem aos critérios para intervenção cirúrgica.
Figura 5 - Desvios de artéria sistêmica-pulmonar. O desvio de Blalock-Taussig conecta a artéria subclávia à artéria pulmonar; o desvio de Waterston conecta a aorta ascendente à artéria pulmonar direita; e o desvio de Potts conecta a aorta descendente à artéria pulmonar esquerda.
Os pacientes submetidos ao reparo da tetralogia de Fallot deverão ser monitorados regularmente quanto à progressão de defeitos residuais, em particular daqueles com regurgitação pulmonar e obstrução de canal condutor. A estenose de ramo arterial pulmonar poderá ser abordada por angioplastia com balão ou stent. A repetição da cirurgia deverá ser considerada em casos de pacientes com DSV residual significativo; pacientes com pressão ventricular direita maior que 2/3 da pressão sistêmica em decorrência de obstrução residual; pacientes com ampliação ventricular direita secundária à regurgitação pulmonar grave e pacientes com tolerância diminuída ao exercício. A substituição da valva pulmonar pode melhorar a função ventricular direita, as arritmias e a capacidade de exercício21. Em adultos, há a possibilidade de realizaçã de uma nova cirurgia com baixo risco. Adultos que tinham canal de condução entre o ventrículo direito e a artéria pulmonar quando crianças poderão apresentar uma relativa obstrução de canal de condução ao longo da vida, conforme as estruturas adjacentes continuam crescendo. Em adição, pode haver uma progressiva estenose de canal ou regurgitação. Os indivíduos com canal de condução do ventrículo direito até a artéria pulmonar ou que passaram por uma substituição prévia da valva pulmonar podem ser candidatos à substituição transcateter da valva pulmonar, que fornece uma valva competente e também alivia a obstrução. A abordagem transcateter está associada a um alto índice de sucesso de procedimento.22 A substituição de valva aórtica ou de raiz aórtica ocasionalmente faz-se necessária, por causa da progressiva regurgitação aórtica.23 As arritmias atriais ocorrem em 10% dos pacientes e foram associadas à regurgitação tricúspide e pulmonar, bem como à idade avançada no momento do reparo.24 As arritmias ventriculares, que são detectadas em 40-50% dos pacientes, foram associadas à idade mais avançada no momento do reparo primário, sobrecarga de volume ventricular direito e prolongamento de QRS. A acentuada ampliação de QRS para mais de 180 ms e a disfunção do ventrículo esquerdo foram identificadas como fatores de risco de morte súbita cardíaca. Nestes casos, é preciso considerar a colocação profilática de um desfibrilador cardíaco implantável.25 A análise cromossômica pode ser útil, se ainda não tiver sido realizada. A tetralogia de Fallot pode estar associada à deleção 22q11, que é uma mutação autossômica dominante e, como tal, tem implicações para os filhos de pais afetados.19
Na forma mais comum de transposição de grandes vasos da base (TGVB) — a destrotransposição (D-TGA) — a aorta surge anteriormente a partir do ventrículo direito, enquanto a artéria pulmonar surge posteriormente a partir do ventrículo esquerdo. As circulações pulmonar e sistêmica estão completamente separadas: o fluxo sanguíneo sistêmico atravessa o lado direito do coração e entra na aorta, enquanto o fluxo sanguíneo pulmonar atravessa o lado esquerdo do coração e entra na artéria pulmonar. A maioria dos pacientes sobreviventes tem CAP e forame oval persistente que permitem a mistura de ambas as circulações. Cerca de 1/3 dos pacientes apresentam anomalias associadas, incluindo DSA e DSV. A obstrução do trato de saída ventricular esquerdo não é incomum. A menos que haja melhora da mistura intracardíaca, a sobrevida além do primeiro ano de vida é incomum.
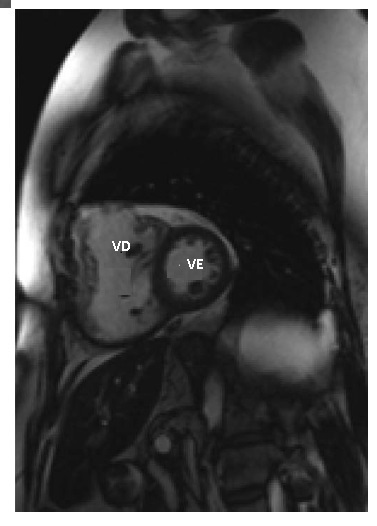
Figura 6 - Imagem de ressonância magnética de um paciente com tetralogia de Fallot reparada e insuficiência de valva pulmonar de longa duração. Note o ventrículo direito (VD) dilatado, em comparação ao ventrículo esquerdo (VE).
|
Tabela 1 Recomendações para substituição de valva pulmonar na tetralogia de Fallot reparada20 |
|
A. RP moderada ou grave (fração regurgitante = 25% por IRM) e pelo menos dois dos seguintes achados: 1. Índice de volume diastólico final VD = 160 mL/m2 (escore z > 5) 2. Índice de volume sistólico final VD = 70 mL/m2 3. Índice de volume sistólico final VE = 65 mL/m2 4. Fração de ejeção = 45% 5. Aneurisma de TSVD 6. Critérios clínicos: intolerância ao exercício, sinais e sintomas de insuficiência cardíaca, medicações para o coração, síncope, taquicardia ventricular contínua B. Outras lesões de relevância hemodinâmica que deflagram o encaminhamento para cirurgia de pacientes com RP moderada a grave C. Os pacientes submetidos ao reparo de TOF com idade > 3 anos deverão ser considerados para substituição de valva pulmonar e na presença de dilação e disfunção VD de menor gravidade.
|
VE = ventricular esquerdo; IRM = imagem de ressonância magnética; RP = regurgitação pulmonar; VD = ventricular direito; TSVD= trato de saída ventricular direito; TOF = tetralogia de Fallot.
A septostomia atrial com balão (procedimento de Rashkind) poderá ser requerida para proporcionar uma mistura melhor ao nível atrial. A cirurgia, inicialmente, consistia em redirecionar o retorno venoso sistêmico para o ventrículo esquerdo e o retorno venoso pulmonar para o ventrículo direito. Estas operações, conhecidas como troca atrial (procedimento de Mustard ou de Senning), que usavam uma tela junto aos átrios, restaurava a circulação fisiológica, mas exigia que o ventrículo direito funcionasse como ventrículo sistêmico. A operação de troca arterial foi substituída pela operação de troca atrial, pelo menos em pacientes com função normal de ambas as valvas semilunares. Na cirurgia de troca arterial, as artérias pulmonar e aorta são, primeiramente, atravessadas acima das valvas semilunares e artérias coronárias, para só então serem trocadas. A aorta é conectada acima da valva pulmônica que surge a partir do ventrículo esquerdo, enquanto a artéria pulmonar é conectada acima da valva aórtica que surge do ventrículo direito. As artérias coronárias são transferidas para a aorta.
Os pacientes com D-TGA e um DSV amplo podem ser submetidos ao procedimento de Rastelli. O fluxo a partir do ventrículo esquerdo é regulado por meio do DSV para a valva aórtica. A artéria pulmonar é dividida e suprassuturada, e um canal condutor é colocado desde o ventrículo direito até a artéria pulmonar, para o fluxo sanguíneo pulmonar.
Os achados físicos estão relacionados à presença de anomalias associadas (i. e., sopros de DSV, estenose pulmônica ou CAP). Os pacientes que recebem tratamento paliativo com procedimento de troca atrial apresentam risco de arritmias e disfunção ventricular direita sistêmica. A disfunção do nodo sinusal é comum, assim como as taquiarritmias atriais, com os resultantes sintomas de fadiga.25 As taquiarritmias atriais, também, foram associadas ao risco aumentado de morte súbita. A disfunção ventricular direita, muitas vezes, se manifesta com sintomas de fadiga e sopro holossistólico de alta frequência por insuficiência tricúspide. É possível que existam problemas adicionais em torno da obstrução ao retorno venoso sistêmico ou pulmonar através das telas de regulação atriais, resultando em ascite/edema periférico ou congestão pulmonar, respectivamente [ver Figura 7] [ver Vídeos 1 a 3].26 Os indivíduos que possuem marca-passo transvenoso apresentam risco aumentado de obstrução da tela de regulação.
Os pacientes que recebedores de tratamento paliativo com troca arterial poderão apresentar sopros relacionados à obstrução do trato de saída residual, produzindo um sopro de ejeção sistólica; um DSA ou DSV residual poderá ser detectado.
Eletrocardiografia. Em pacientes com troca atrial, o ECG mostra desvio de eixo à direita e HVD. Em pacientes com troca arterial, o ECG pode ser normal, desde que o fluxo sanguíneo coronariano não esteja comprometido.
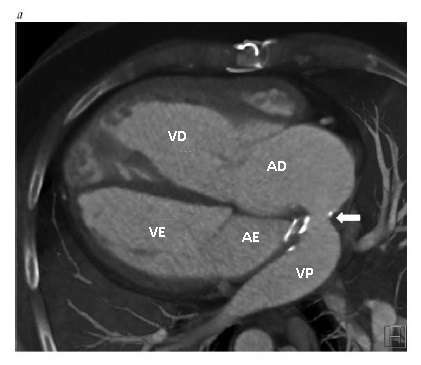
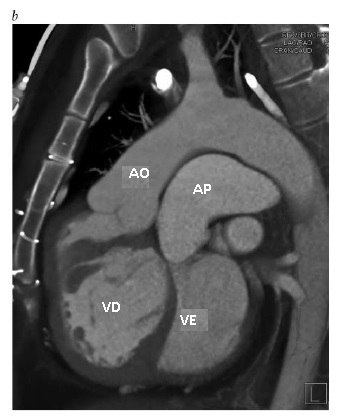
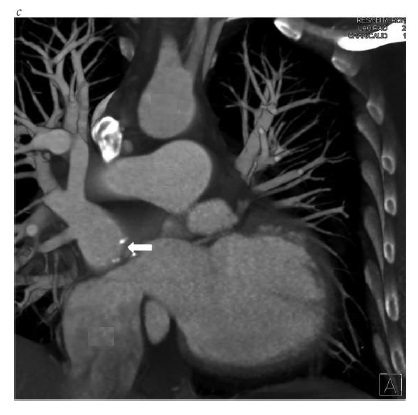
Figura 7 - Imagens de tomografia computadorizada de um paciente que recebeu tratamento paliativo de troca atrial para transposição de grandes artérias e obstruções múltiplas de tela de regulação. (a), Vista axial, mostrando as veias pulmonares com tela de regulação dentro do átrio direito nativo, que drena no interior do ventrículo direito morfológico que, por sua vez, se conecta à aorta. O retorno venoso sistêmico drena dentro do átrio esquerdo e, então, dentro do ventrículo esquerdo morfológico. (b), Vista sagital, mostrando a localização da aorta anterior à artéria pulmonar. Note que a aorta surge do ventrículo direito morfológico, enquanto a artéria pulmonar surge do ventrículo esquerdo morfológico. A seta branca identifica a estenose da tela de regulação venosa pulmonar com calcificação de material de enxerto sintético. (c), Vista coronal, mostrando a VCI dilatada drenando através da tela de regulação para dentro do ventrículo esquerdo morfológico. A seta branca identifica a estenose da tela de regulação venosa pulmonar com calcificação de material de enxerto sintético. Há ainda uma grave estenose associada da tela de regulação venosa sistêmica superior. AO = aorta, VCI = veia cava inferior, AE = átrio esquerdo, VE = ventrículo esquerdo subpulmonar, AP = artéria pulmonar, VP = veia pulmonar, AD = átrio direito, VD = ventrículo direito sistêmico.
Exames radiológicos. Os pacientes submetidos ao procedimento de troca atrial, geralmente, apresentam cardiomegalia a partir de um ventrículo direito dilatado ao exame de raio x de torax, enquanto a artéria pulmonar pode mostrar um fluxo preferencial para o pulmão direito. A angiografia por ressonância magnética é útil para identificar os defeitos residuais e medir a função ventricular direita. Os pacientes com reparo por troca arterial tendem a ter um coração de tamanho normal. Um fluxo sanguíneo pulmonar assimétrico implica em obstrução de ramo arterial pulmonar.
Ecocardiografia. A ecocardiografia é usada na avaliação de defeitos residuais associados: função ventricular direita deprimida; regurgitação tricúspide progressiva (valva atrioventricular sistêmica); obstrução do trato de saída ventricular esquerdo; DSV residual ou anormalidades de perfusão arterial coronariana.
Após a troca atrial, a perspectiva a longo prazo é muito boa, com mais de 95% dos pacientes sem nenhum sintoma significativo. Entretanto, estes pacientes precisam ser monitorados quanto à ampliação progressiva do ventrículo direito e regurgitação tricúspide, levando à disfunção ventricular que, por sua vez, é problemática e aumentará conforme a população de pacientes envelhece. Estes pacientes requerem procedimentos cirúrgicos paliativos, agressivos ou transplante cardíaco, caso a terapia médica seja inefetiva. As arritmias atriais, incluindo a síndrome do nó sinoatrial e o flutter atrial, são comuns.25 A morte súbita pode ocorrer em 6-17% dos pacientes.27 A tela de regulação atrial pode causar obstrução venosa sistêmica ou pulmonar, que é abordada com uma nova operação ou por angioplastia com balão e colocação de stent.
O prognóstico a longo prazo de pacientes com troca arterial é menos conhecido, porém as arritmias são consideradas menos frequentes e secundárias a imperfeições do procedimento operatório.28 Caso surja alguma preocupação, os pacientes devem ser submetidos a exames de medicina nuclear ou testes de estresse para monitoramento de perfusão coronariana inadequada secundária a anormalidades de reimplantação da artéria coronária. Poderá haver estenose da artéria pulmonar principal ou de ramos arteriais pulmonares (a complicação mais comum), ou ainda estenose em sítios de anastomose aórtica. As complicações do procedimento de Rastelli incluem a obstrução subaórtica (obstrução de tela de regulação ou DSV), estenose de canal condutor (com ou sem regurgitação), vazamento de tela de regulação e estenose de ramo arterial pulmonar. Os defeitos residuais significativos poderão requerer intervenção.26 Os pacientes que tiveram um canal condutor implantado, também, poderão ser candidatos à substituição transcateter de valva pulmonar para abordagem da disfunção de canal.
Um único ventrículo funcional pode resultar de defeitos como a síndrome do coração esquerdo hipoplásico (atresia aórtica, atresia mitral ou ambas), atresia tricúspide, atresia pulmonar com septo ventricular intacto ou DSAV não equilibrado, causando desenvolvimento preferencial de apenas um ventrículo.
Na infância, a manifestação inicial de um coração univentricular poderá incluir uma cianose grave associada a uma acentuada diminuição do fluxo sanguíneo pulmonar; cianose leve e insuficiência cardíaca associada a um fluxo sanguíneo acentuadamente diminuído e mistura intracardíaca de circulações; ou fluxos sanguíneos sistêmico e pulmonar quase equilibrados e cianose leve.29 Os pacientes que sobrevivem até a fase adulta são submetidos a, pelo menos, um procedimento cirúrgico paliativo. Entre estes, estão o procedimento de Norwood, anastomose cavopulmonar bidirecional parcial (procedimento de Glenn) e anastomose cavopulmonar completa (procedimento de Fontan). As operações, hoje, são tipicamente realizadas com uma abordagem de estágios ao longo da infância.
Paliação neonatal. Os procedimentos paliativos neonatais variam; todos proporcionam uma fonte estável de fluxo sanguíneo pulmonar, em geral, por meio de um desvio a partir da aorta ou de seus ramos para as artérias pulmonares (desvio central ou desvio de Blalock-Taussig modificado). A operação de Norwood para síndrome do coração esquerdo hipoplásico é feita pouco após o nascimento e estabelece uma saída única no coração ao anastomosar a aorta ascendente hipoplásica à artéria pulmonar principal, produzindo uma neoaorta e conectando a artéria pulmonar distal a um desvio, usualmente um desvio de Blalock-Taussigshunt modificado [ver Figura 5] ou um desvio de Sano que consiste em um canal condutor estendendo-se do ventrículo direito até as artérias pulmonares [ver Figura 8a]. Uma septectomia atrial tipicamente é necessária para permitir a mistura completa ao nível atrial.
Procedimento de Glenn (anastomose cavopulmonar superior)
O procedimento bidirecional de Glenn envolve a anastomose da VCS à artéria pulmonar. Inclui a retirada de um desvio previamente colocado e o reparo de qualquer estenose de ramo arterial pulmonar [ver Figura 8b]. O termo “bidirecional” se refere ao fato da artéria pulmonar direita permanecer em continuidade com a artéria pulmonar esquerda. Isto contrasta com o procedimento de Glenn clássico, que envolve a anastomose da VCS à artéria pulmonar direita, desconectada das artérias pulmonares principal e esquerda. O procedimento bidirecional de Glenn, atualmente, é comumente realizado aos 4-6 meses de idade.
Procedimento de Fontan (anastomose cavopulmonar total)
O procedimento de Fontan é o procedimento paliativo final, que promove uma conexão direta entre o fluxo oriundo da veia cava inferior (VCI) com o circuito pulmonar. Inicialmente, em vez das intervenções em estágios, uma única operação envolvendo a fixação do átrio direito à artéria pulmonar ou ao trato de saída ventricular direito era realizada em pacientes com mais de 4 anos de idade. A prática corrente consiste em direcionar o retorno venoso sistêmico ao circuito pulmonar por meio de procedimento estadiados: no primeiro estágio, a VCS é anastomosada à artéria pulmonar durante a infância (procedimento de Glenn); no segundo, o fluxo da VCI é direcionado para as artérias pulmonares aos 2-3 anos de idade (acabamento de Fontan). O fluxo da VCI direcionado para a artéria pulmonar, seja por um túnel lateral instalado ao longo do átrio direito [Figura 8c] ou por um canal condutor extracardíaco conectando a VCI diretamente à artéria pulmonar [Figura 8d]. Com qualquer uma destas vias de fluxo, uma pequena comunicação (fenestração) poderá ser criada entre o canal condutor de fluxo sanguíneo caval e o átrio esquerdo funcional, com a finalidade de descomprimir o circuito de Fontan no evento de uma pressão venosa central elevada. O fluxo sanguíneo pulmonar é conseguido via retorno venoso passivo, sem auxílio de uma câmara bombeadora ventricular. Qualquer alteração discreta na resistência ou pressão pulmonar comprometerá a adequação do fluxo sanguíneo pulmonar. Portanto, é ideal a eliminação de qualquer tipo de estenose de ramo arterial pulmonar.
As características clínicas são variáveis; alguns pacientes podem obter uma paliação efetiva, com saturação de oxigênio quase normal, níveis de atividade aceitáveis e achados negligíveis ao exame cardíaco. Outros apresentarão insuficiência cardíaca progressiva conforme o ventrículo único (em especial, se este estiver anastomosado ao ventrículo direito) sucumbe à pressão aumentada e à sobrecarga de volume secundária à progressiva regurgitação da valva atrioventricular e disfunção do miocárdio. As arritmias atrial e ventricular são comuns e o lento fluxo sanguíneo pulmonar poderá predispor o desenvolvimento de trombose in situ e a embolia pulmonar que, por sua vez, obstruirão o fluxo sanguíneo pulmonar elevando a pressão arterial pulmonar.
Eletrocardiografia. Os achados de ECG são bastantes variáveis e poderão incluir aumento da amplitude atrial ou ventricular; desvio de eixo; anomalias de condução; e arritmias. Esta variabilidade está relacionada às variações na anatomia subjacente que é amenizada em uma via comum única.
Figura 8 - Estágios do reparo de ventrículos únicos funcionais (ver detalhes no texto). (a) Procedimento de Norwood; (b) anastomose cavopulmonar parcial bidirecional (procedimento de Glenn); (c) anastomose cavopulmonar total de túnel lateral; (d) anastomose cavopulmonar total de canal condutor extracardíaco.
Exames radiológicos. O raio x de torax poderá mostrar uma cardiomegalia progressiva. As marcas vasculares pulmonares poderão ser desiguais, indicando a estenose de um ou mais ramos arteriais pulmonares. A IRM com angiografia poderá revelar áreas de estenose de ramo arterial pulmonar e alterações progressivas no tamanho da câmara e na função ventricular. O cateterismo cardíaco, também, poderá fornecer imagens do circuito de Fontan.
Ecocardiografia. Os exames ecocardiográficos são realizados com a finalidade de seguir a progressão da regurgitação da valva atrioventricular, alargamento ventricular e disfunção, bem como detectar a chamada fumaça ou coágulos no circuito venoso sistêmico-arterial pulmonar, que são consequência do fluxo sanguíneo lento e passivo. Poderá ser difícil a obtenção de uma imagem do ramo de artérias pulmonares por ecocardiografia e, neste caso, recomenda-se uma tentativa de obtenção de imagens adicionais das artérias pulmonares.
Após a correção cirúrgica, os pacientes apresentam limitações significativas da tolerância ao exercício, por contarem, apenas, com o fluxo sanguíneo pulmonar passivo que não aumenta maximamente com o esforço. As arritmias pós-operatórias são comuns. Tais arritmias poderão requerer tratamento médico, pois as técnicas de ablação por radiofrequência poderão ser limitadas pela falta de acesso transvenoso ao tecido atrial nativo após o acabamento de Fontan. Os sítios prévios de implantação de desvio nas artérias pulmonares particularmente apresentam alto risco de estenose. As intervenções transcateter poderão ser necessárias para a abordagem de estenoses de ramo arterial pulmonar, vazamentos de tela de regulação, colaterais venovenosos ou colaterais arteriovenosos. Como os vazamentos de tela de regulação e vasos colaterais poderão resultar em potencial de desvio da direita-esquerda, a introdução de ar em linhas intravenosas deverá ser evitada. A necessidade de uma nova operação após um procedimento de Fontan é infrequente e sua indicação mais comum é a colocação de marca-passo. A enteropatia perdedora de proteína (EPP) é um grave problema subsequente à operação de Fontan, que está associado a uma taxa de sobrevida de 5 anos da ordem de 59%.30 Sua causa é indeterminada, mas, provavelmente, está relacionada a pressões sistêmica venosa e de ducto torácico aumentadas. É possível que, também, exista um componente autoimune ou alérgico local na parede intestinal. A EPP é caracterizada por edema periférico, má absorção e níveis séricos de proteína baixos. As complicações se tornam menos frequentes com a cirurgia estadiada e o fornecimento de fenestração. Alguns pacientes de idade mais avançada poderão ser beneficiados pela conversão do procedimento de Fontan clássico em uma anastomose total cavoarterial pulmonar. Em casos de insuficiência ventricular sistêmica ou EPP intratável, o transplante cardíaco poderá ser necessário.31
Esta anomalia incomum da valva tricúspide consiste na aderência dos folhetos posterior e septal ao miocárdio, causando a descida do ânulo funcional para o ápice do ventrículo direito e o alargamento do folheto anterior. O resultado final é uma atrialização de parte do ventrículo direito, com resultante insuficiência tricúspide. Em pacientes que manifestam a condição antecipadamente ao longo da vida, a anomalia de Ebstein, muitas vezes, é encontrada associada a outros defeitos, incluindo DSA e estenose pulmônica. As vias acessórias e evidência clínica de pré-excitação não são incomuns, enquanto as arritmias são os achados mais comumente encontrados em adultos.
A anomalia de Ebstein, por sua vez, poderá tornar-se clinicamente evidente em qualquer idade. A história natural desta lesão varia de morte precoce à sobrevida- até a fase adulta- sem cirurgia, dependendo do grau de regurgitação e da ocorrência de arritmias significativas. Poderá haver cianose, tanto em neonatos como em adultos, secundária ao desvio direita-esquerda ao nível atrial. Os pacientes adultos poderão queixar-se de fadiga, falta de ar, palpitações ou síncope. À auscultação, um sopro de regurgitação tricúspide é evidente e, frequentemente, está associado a um ritmo de galope, múltiplos sons de ejeção sistólica e um segundo som amplamente separado.
Eletrocardiografia. Os achados de ECG são bastante variáveis. O intervalo PR poderá estar normal, curto com pré-excitação, ou prolongado. O eixo poderá ser superior ou à direita, com ou sem bloqueio de feixe à direita. Poderá haver evidência de ampliação atrial direita. As arritmias são detectadas em quase metade dos casos.32
Exames radiológicos. O raio x torácico poderá mostrar cariomegalia com ampliação atrial direita. Não há necessidade de cateterismo cardíaco, a menos que exista alguma preocupação relacionada à arteriopatia coronariana ou diante da necessidade de avaliação eletrofisiológica, e possível ablação por radiofrequência.
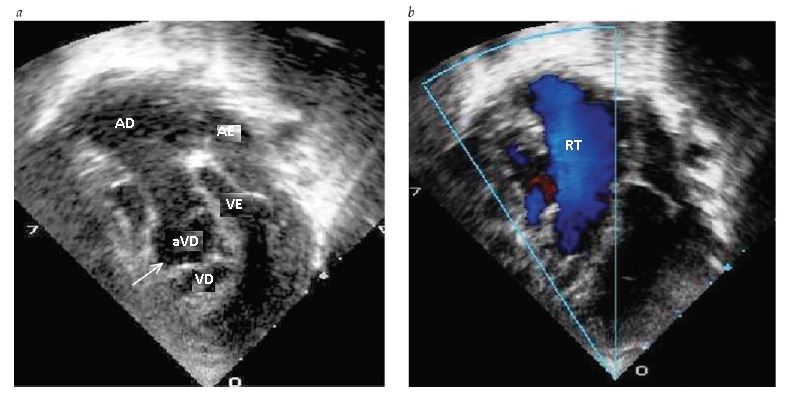
Ecocardiografia. A ecocardiografia poderá trazer a confirmação do diagnóstico e do grau de deslocamento da valva tricúspide, que poderá variar de um leve encostar do folheto septal a um grave deslocamento apical, caracterizandoa gravidade da regurgitação tricúspide [ver Figura 9]. O folheto anterior é amplo e semelhante a uma vela, podendo produzir obstrução da saída ventricular direita. O septo atrial deverá ser avaliado quanto ao tamanho do defeito e magnitude do desvio.
A cirurgia é recomendada para pacientes com insuficiência cardíaca sintomática e cardiomegalia, cianose ou arritmias. A valvuloplastia tricúspide é preferida à substituição de valva. Não recomenda-se a cirurgia para pacientes assintomáticos, contudo uma intervenção deverá ser considerada se houver deterioração funcional, antes do desenvolvimento de sintomas significativos.33
Uma séria complicação dos desvios de esquerda-direita de longa duração nos átrios, ventrículos ou grandes artérias é o desenvolvimento de uma hipertensão arterial pulmonar irreversível e grave denominada síndrome de Eisenmenger.
Normalmente, a resistência vascular pulmonar é substancialmente menor do que a resistência vascular sistêmica. Isto facilita a ocorrência de um desvio significativo de esquerda-direita a partir de amplas comunicações intracardíacas ou grandes artérias. À medida que a resistência vascular pulmonar aumenta, a resistência ao fluxo para dentro da circulação pulmonar eventualmente excederá a resistência ao fluxo para dentro da circulação sistêmica, levando ao desvio de direita-esquerda. Isto resultará em graus variáveis de cianose e outros achados físicos de hipertensão pulmonar. Diferente dos pacientes com policitemia vera ou policitemia por doença pulmonar obstrutiva crônica, os pacientes com síndrome de Eisenmenger, muitas vezes, requerem hematócritos ao redor dos 60 ou, até mesmo,no início dos 70 anos, para distribuir oxigênio suficiente aos tecidos e evitar sintomas isquêmicos.
Figura 9 - Ecocardiogramas de um paciente com anomalia de Ebstein. (a) Vista apical das quatro câmaras. A seta indica um deslocamento apical do folheto tricúspide. (b) Imagem colorida de fluxo com Doppler, mostrando uma grave regurgitação tricúspide (RT), originária das profundezas do ventrículo direito, a partir do folheto da valva tricúspide deslocado. aRV = ventrículo direito “atrializado”; AE = átrio esquerdo; VE = ventrículo esquerdo; AD = átrio direito; VD = ventrículo direito.
Os pacientes com síndrome de Eisenmenger poderão ser assintomáticos, exceto pela cianose. Eventualmente, muitos pacientes notarão uma diminuição da tolerância ao exercício e desconforto torácico, com frequência reminiscente de angina de peito. Quando a eritrocitose secundária atinge níveis graves, os pacientes poderão desenvolver sintomas de hiperviscosidade, incluindo perturbações visuais, cefaleias e outras queixas.
O exame físico de um paciente com síndrome de Eisenmenger revelará manifestações de hipertensão pulmonar, incluindo um sonoro componente pulmonar da segunda bulha cardíaca e o sopro diastólico alto de regurgitação pulmonar de alta pressão (sopro de Graham Steell). Entre os achados adicionais, estão a cianose, baqueteamento e a elevação ou levantamento ventricular direito.
Eletrocardiografia. O ECG mostra desvio de eixo à direita e HVD, exibida em forma de ondas R altas e anormalias ST-T em V1 a V3.
Exames radiológicos. O raio x de torax mostrará artérias pulmonares centrais ampliadas com poda arterial periférica. A cardiomegalia com ampliação de câmara específica refletirá o defeito subjacente. O cateterismo cardíaco de lado direito, muitas vezes, faz-se necessário para avaliar a resistência e pressão arterial pulmonar.
Ecocardiografia. A ecocardiografia identificará e quantificará o desvio cardíaco subjacente, além de fornecer uma estimativa das pressões cardíacas do lado direito.
Os pacientes com síndrome de Eisenmenger poderão viver por décadas após o diagnóstico.34 Alternativamente, poderá ocorrer morte súbita por arritmias ventriculares. Como a resistência pulmonar é alta e fixa nestes pacientes, é preciso evitar situações que possam levar a quedas repentinas da resistência vascular sistêmica, que exacerbariam o desvio de direita-esquerda, por vezes de modo potencialmente fatal. Isto implicaria evitar ambientes excessivamente quentes e desidratação. Adicionalmente, é preciso ter cuidado durante a anestesia ou com o uso de fármacos vasodilatadores. A gravidez é outro estado em que a resistência vascular sistêmica cai. Por isso, a gravidez é extremamente perigosa para uma mãe com hipertensão pulmonar, bem como para o feto, sendo assim absolutamente contraindicada na síndrome de Eisenmenger. A deficiência de ferro, quando presente, deverá ser tratada. Somente em raros casos, a flebotomia será necessária para aliviar os sintomas de hiperviscosidade. Agentes como a prostaciclina, antagonistas da endotelina ou inibidores de 5-fosfodiesterase são usados para diminuir a resistência arteriolar pulmonar.35–37 Transplantes de coração-pulmão ou apenas de pulmão foram realizados com êxito em alguns pacientes com síndrome de Eisenmenger.38
Os anticoncepcionais contendo estrógeno apresentam elevada eficácia contraceptiva, mas estão associados ao risco aumentado de trombose arterial e venosa. Portanto, os anticoncepcionais contendo apenas progestina são favoráveis para as mulheres com preocupações relacionadas à trombose. Uma forma de ação duradoura pode melhorar a eficácia.39 A seleção de métodos de contracepção deverá ser feita mediante consultas conjuntas a um obstetra e a um cardiologista.
Considerando a sobrevida crescente de indivíduos que nascem com CC, o número de mulheres em idade fértil com CCs está aumentando. As malformações congênitas são as formas mais comuns de cardiopatia em gestantes. As alterações fisiológicas, ocorridas durante a gestação, resultam no aumento gradual do volume sanguíneo e do débito cardíaco, bem como na diminuição da resistência vascular periférica. Estas alterações são revertidas de forma relativamente rápida, após o parto. Contudo, estes ajustes poderão ser mal tolerados em gestantes com diminuição preexistente da função cardíaca.40 As gestantes com arritmias poderão apresentar exacerbações durante a gestação e poderão ser beneficiadas por uma terapia de ablação transcateter instituída antes da concepção. Algumas condições cardíacas estão associadas a um risco extremamente alto de mortalidade materna ou morbidade grave, que faz com que a gravidez seja contraindicada [ver Tabela 2].39 Por outro lado, muitas mulheres com CC conseguem tolerar a gestação.
As mulheres com CC podem ser tratadas com medicações anti-hipertensivas ou anticoagulantes que são contraindicadas durante a gestação. Por este motivo, o aconselhamento pré-concepção deverá ser iniciado durante a adolescência, para que as medicações possam ser ajustadas antes da concepção. De um modo geral, os resultados adversos fetais e neonatais ocorrem em 15-39% das gestações.40 Entre as mães com cianose significativa (saturação de oxigênio arterial <85%), excluindo aquelas com síndrome de Eisenmenger, apenas 12% das gestações resultam no nascimento de bebês vivos.41 As mães com CC são mais propensas a terem filhos com CC. Este risco aumenta ainda mais entre as mães com obstrução cardíaca de lado esquerdo ou com etiologia genética comprovada, como a deleção cromossômica 22q11. Os ecocardiogramas fetais poderão ser valiosos para a triagem de CCs em mães com CC, nas gestações em que outras anomalias foram identificadas, ou, ainda, para outras indicações [ver Tabela 3]. Os ecocardiogramas fetais ,idealmente, são realizados entre 18 e 22 semanas de gestação. Entretanto, as anomalias venosas pulmonares e a coarctação da aorta são de difícil detecção in utero. Veja mais informações adiante, na barra lateral Fontes Selecionadas Sobre Cardiopatia Congênita na Internet.
|
Tabela 2 Condições em que a gravidez é de alto risco39 |
|
a. Hipertensão arterial pulmonar de qualquer causa b. Disfunção ventricular sistêmica grave NYHA III–IV ou FEVE = 30% c. Miocardiopatia periparto prévia com qualquer comprometimento residual da função ventricular esquerda d. Obstrução cardíaca de lado esquerdo grave e. Síndrome de Marfan com dilatação da aorta > 40 mm |
|
Tabela 3 Indicações cardíacas para ecocardiografia fetal42–44 |
|
Indicações fetais 1. Bradi- ou taquiarritmias 2. Vista anormal das quatro câmaras ou do eixo cardíaco Indicações familiares 1. Parente de primeiro grau com defeito cardíaco congênito (mãe, irmão ou pai) 2. Síndromes mendelianas (esclerose tuberosa, síndrome de Noonan, síndrome de DiGeorge) |
|
Fontes Selecionadas Sobre Cardiopatia Congênita na Internet |
|
International Society for Adult Congenital Heart Disease (ISACHD) http://www.isachd.org Fontes profissionais, informações para pacientes e newsletter Canadian Adult Congenital Heart Network (CACH) http://www.cachnet.org Informação para médicos e pacientes Grown Up Congenital Heart Patients Association (GUCH) http://www.guch.org.uk/ Site do Reino Unido que fornece informação e suporte para os pacientes e suas famílias PediHeartNet http://www.pediheart.net Informações para profissionais e pacientes, lista de correspondência Adult Congenital Heart Association http://www.achaheart.org Informações para profissionais e pacientes Adult Congenital & Cardiovascular Genetics Center http://www.umn-accgc.congenital.org Informação para pacientes, em inglês e espanhol Medtronic Melody® Transcatheter Pulmonary Valve http://www.medtronic.com/melody/procedure.html Animações e imagens de fluoroscopia de preparações de valva |
Os autores não possuem relações comerciais com os fabricantes de produtos ou prestadores de serviços discutidos nesta subseção.
1.Marelli AJ, Mackie AS, Ionescu-Ittu R, et al. Congenital heart disease in the general population: changing prevalence and age distribution. Circulation 2007;115:163–72.
2.Webb G, Gatzoulis MA. Atrial septal defects in the adult: recent progress and overview. Circulation 2006;114:1645– 53.
3.Driscoll DJ. Left-to-right shunt lesions. Pediatr Clin North Am1999;46:355–68, x.
4.Inglessis I, Landzberg MJ. Interventional catheterization in adult congenital heart disease. Circulation 2007;115:1622– 33.
5. Fischer G, Stieh J, Uebing A, et al. Experience with trans- catheter closure of secundum atrial septal defects using the Amplatzer septal occluder: a single centre study in 236 consecutive patients. Heart 2003;89:199–204.
6.Gatzoulis MA, Freeman MA, Siu SC, et al. Atrial arrhythmia after surgical closure of atrial septal defects in adults. N Engl J Med 1999;340:839–46.
7.Divekar A, Gaamangwe T, Shaikh N, et al. Cardiac perforation after device closure of atrial septal defects with the Amplatzer septal occluder. J Am Coll Cardiol 2005;45: 1213–8.
8.Suzuki K, Yamaki S, Mimori S, et al. Pulmonary vascular disease in Down’s syndrome with complete atrioventricu- lar septal defect. Am J Cardiol 2000;86:434–7.
9.Goyal VS, Fulwani MC, Ramakantan R, et al. Follow-up after coil closure of patent ductus arteriosus. Am J Cardiol1999;83:463–6, A10.
10.Lewin MB, Otto CM. The bicuspid aortic valve: adverse outcomes from infancy to old age. Circulation 2005;111:832–4.
11.Roman MJ, Devereux RB, Kramer-Fox R, O’Loughlin J.Two-dimensional echocardiographic aortic root dimensions in normal children and adults. Am J Cardiol 1989;64: 507–12.
12.Stanger P, Cassidy SC, Girod DA, et al. Balloon pulmonary valvuloplasty: results of the valvuloplasty and angioplasty of congenital anomalies registry. Am J Cardiol 1990;65: 775–83.
13.Bashore TM. Adult congenital heart disease: right ventricular out?ow tract lesions. Circulation 2007;115:1933–47.
14.Oliver JM, Gallego P, Gonzalez A, et al. Risk factors for aortic complications in adults with coarctation of the aorta. J Am Coll Cardiol 2004;44:1641–7.
15.Forbes TJ, Kim DW, Du W, et al. Comparison of surgical, stent, and balloon angioplasty treatment of native coarctation of the aorta: an observational study by the CCISC (Congenital Cardiovascular Interventional Study Consortium). J Am Coll Cardiol 2011;58:2664–74.
16.Moss AJ, Allen HD. Moss and Adams’ heart disease in infants, children, and adolescents: including the fetus and young adult. 7th ed. Philadelphia: Wolters Kluwer Health/ Lippincott Williams & Wilkins; 2008.
17.Ratliff HL, Yousufuddin M, Lieving WR, et al. Persistent left superior vena cava: case reports and clinical implica- tions. Int J Cardiol 2006;113:242–6.
18.Brickner ME, Hillis LD, Lange RA. Congenital heart disease in adults. Second of two parts. N Engl J Med 2000;342:334– 42.
19.Goldmuntz E, Clark BJ, Mitchell LE, et al. Frequency of 22q11 deletions in patients with conotruncal defects. J Am Coll Cardiol1998;32:492–8.
20.Geva T. Indications and timing of pulmonary valve replacement after tetralogy of Fallot repair. Semin Thorac Cardiovasc Surg Pediatr Cardiac Surg Annu 2006;9:11–22.
21.Henkens IR, van Straten A, Schalij MJ, et al. Predicting outcome of pulmonary valve replacement in adult tetralogy of Fallot patients. Ann Thorac Surg 2007;83:907–11.
22.McElhinney DB, Hellenbrand WE, Zahn EM, et al. Short- andmedium-term outcomes after transcatheter pulmonary valve placement in the expanded multicenter US Melody Valve Trial. Circulation 2010;122:507–16.
23.Niwa K, Siu SC, Webb GD, Gatzoulis MA. Progressive aortic root dilatation in adults late after repair of tetralogy of Fallot. Circulation 2002;106:1374–8.
24.Gatzoulis MA, Balaji S, Webber SA, et al. Risk factors for arrhythmia and sudden cardiac death late after repair of tetralogy of Fallot: a multicentre study. Lancet 2000;356:975–81.
25.Dos L, Teruel L, Ferreira IJ, et al. Late outcome of senning and mustard procedures for correction of transposition of the great arteries. Heart 2005;91:652–6.
26.Warnes CA. Transposition of the great arteries. Circulation2006;114:2699–709.
27.Wilson NJ, Clarkson PM, Barratt-Boyes BG, et al. Long-termoutcome after the mustard repair for simple transposition of the great arteries. 28-year follow-up. J Am Coll Cardiol1998;32:758–65.
28.Hutter PA, Kreb DL, Mantel SF, et al. Twenty-?ve years’ experience with the arterial switch operation. J Thorac Cardiovasc Surg 2002;124:790–7.
29.Khairy P, Poirier N, Mercier LA. Univentricular heart. Circulation 2007;115:800–12.
30.Mertens L, Hagler DJ, Sauer U, et al. Protein-losing enteropathy after the Fontan operation: an international multicenter study. PLE Study Group. J Thorac Cardiovasc Surg 1998;115:1063–73.
31.Therrien J, Warnes C, Daliento L, et al. Canadian Cardio- vascular Society Consensus Conference 2001 update: recommendations for the management of adults with con- genital heart disease part III. Can J Cardiol 2001;17:1135– 58.
32.Celermajer DS, Bull C, Till JA, et al. Ebstein’s anomaly: presentation and outcome from fetus to adult. J Am Coll Cardiol1994;23:170–6.
33.Attenhofer Jost CH, Connolly HM, Dearani JA, et al. Ebstein’s anomaly. Circulation 2007;115:277–85.
34.Oya H, Nagaya N, Uematsu M, et al. Poor prognosis and related factors in adults with Eisenmenger syndrome. Am Heart J2002;143:739–44.
35.Mukhopadhyay S, Sharma M, Ramakrishnan S, et al.Phosphodiesterase-5 inhibitor in Eisenmenger syndrome: a preliminary observational study. Circulation 2006;114:1807–10.
36.Galie N, Beghetti M, Gatzoulis MA, et al. Bosentan therapy in patients with Eisenmenger syndrome: a multicenter,double-blind, randomized, placebo-controlled study. Circulation2006;114:48–54.
37.Diller GP, Gatzoulis MA. Pulmonary vascular disease in adults with congenital heart disease. Circulation 2007;115: 1039–50.
38.Franke UF, Wahlers T, Wittwer T, et al. Heart-lungtransplantation is the method of choice in the treatment of patients with end-stage pulmonary hypertension. Transplant Proc 2002;34:2181–2.
39.Thorne S, MacGregor A, Nelson-Piercy C. Risks of contra- ception and pregnancy in heart disease. Heart 2006;92: 1520–5.
40.Fernandes SM, Arendt KW, Landzberg MJ, et al. Pregnant women with congenital heart disease: cardiac, anesthetic and obstetrical implications. Expert Rev Cardiovasc Ther2010;8:439–48.
41. Presbitero P, Somerville J, Stone S, et al. Pregnancy in cyanotic congenital heart disease. Outcome of mother and fetus. Circulation 1994;89:2673–6.
42.Small M, Copel JA. Indications for fetal echocardiography. Pediatr Cardiol 2004;25:210–22.
43. Rychik J, Ayres N, Cuneo B, et al. American Society of Echocardiography guidelines and standards for performance of the fetal echocardiogram. J Am Soc Echocardiogr 2004;17:803–10.
44. AIUM practice guideline for the performance of fetal echocardiography. J Ultrasound Med 2011;30:127–36.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.