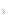 (Carregando Índice)... (Carregando Índice)... |
Última revisão: 09/01/2018
Comentários de assinantes: 0
|
Artigo original: Kantarjian, M, H, MD. Versovsek. Essential Thrombocythemia and Myelofibrosis, SAM. [The original English language work has been published by DECKER INTELLECTUAL PROPERTIES INC. Hamilton, Ontario, Canada. Copyright © 2015 Decker Intellectual Properties Inc. All Rights Reserved.] Tradução: Paulo Henrique Machado. Revisão técnica: Dr. Lucas Santos Zambon. |
Hagop M. Kantarjian, MD
Professor e Diretor do Departamento de Leucemia da The University of Texas MD Anderson Cancer Center (Houston, TX).
Srdan Verstovsek, MD, PhD
Professor do Departamento de Leucemia da The University of Texas MD Anderson Cancer Center (Houston, TX).
Resumo
Esta revisão apresenta detalhes sobre os dois maiores distúrbios clonais das células-tronco: trombocitemia essencial (TE) e mielofibrose (MF). A TE se caracteriza pela proliferação sustentada de megacariócitos resultando em trombocitose no sangue periférico. A mielofibrose primária (MFP) está associada a condições como hematopoiese extramedular; esplenomegalia, um quadro de sangue leucoeritroblástico; e a vários graus de fibrose medular com hiperplasia megacariocítica e atipia acentuadas. Serão contempladas a epidemiologia, a etiologia/genética, a patogênese, o diagnóstico (incluindo manifestações clínicas e exames laboratoriais), os diagnósticos diferenciais, o gerenciamento e o prognóstico de cada distúrbio. Esta revisão inclui também opções para o tratamento de MF, incluindo interferon a, inibidores da JAK e transplante alogênico de células-tronco (TACT); o TACT ainda é o único tratamento curativo para MF. As figuras mostram algoritmos de tratamento para TE e MF. Os quadros apresentam uma lista dos critérios atuais da Organização Mundial da Saúde (OMS) para diagnósticos de TE e MF, orientações do International Working Group for Myelofibrosis Research and Treatment (IWG-MRT) para diagnósticos no período pós-TE/MF, fatores prognósticos no International Prognostic Score for ET (IPSET) e no IPSET-thrombosis, sistema de pontuação prognóstica para MF e atividade clínica dos inibidores da JAK2.
HEMATOLOGIA
Trombocitemia Essencial
A trombocitemia essencial (TE) é um distúrbio clonal de células-tronco que se caracteriza pela proliferação sustentada de megacariócitos, resultando em trombocitose no sangue periférico.
Epidemiologia
A incidência de TE é de, aproximadamente, 2,2 a cada 100 mil pessoas. A idade média no momento do diagnóstico varia de 55 a 60 anos. A TE afeta mais as mulheres do que os homens, em uma proporção de 2:1.1
Etiologia/Genética
Ainda não se conhece a etiologia precisa da TE. Embora não se acredite que seja hereditária, os parentes de pacientes com neoplasma mieloproliferativo (NMP) têm uma probabilidade sete vezes maior de desenvolver esse tipo de doença,2 sugerindo a hipótese de uma predisposição genética para a TE.
Foram identificados dois tipos de predisposição genética em NMPs, incluindo a TE. Um deles é a predisposição associada ao haplótico 46/1 da quinase 2 de Janus (JAK2, do inglês Janus kinase 2), que aumenta o risco de abrigar mutações no gene da quinase 2 de Janus.3,4 Aparentemente, em casos raros, há uma predisposição familiar com um padrão autossômico dominante de herança.5 Entretanto, o gene ou genes responsáveis pela herança familiar não são conhecidos.
As anormalidades cariotípicas são raras nos casos de TE ? apenas 5 a 10% de pacientes apresentam um cariótipo anormal ? e, entre elas, as mais comuns são trissomia 8 ou 9, deleção do 13q e deleção do 20q.6 Até 90% dos pacientes com TE apresentam mutações em um entre três genes que, provavelmente, agem como desencadeadores do processo da doença. A mutação mais comum, encontrada em 50 a 60% de pacientes com TE, se localiza no gene JAK2V617F da quinase 2 de Janus7?9
Um adicional de 5% de pacientes abriga mutações no gene que codifica o receptor da trombopoietina (MPL), MPLW515L/K.10 Mais recentemente, mutações no gene calreticulin (CALR) foram encontradas em até 70% de pacientes que eram negativos para as mutações JAK2 ou MPL (25% do total de casos de TE).11,12 Essas três mutações são exclusivas entre si, sendo que todas elas ativam a rota de sinalização intracelular JAK-STAT (transdutor de sinal e ativador de transcrição) que, em geral, é o problema biológico subjacente nos NMPs.
Patogênese
As células hematopoiéticas progenitoras na TE são francamente responsivas à ação das citocinas e dos hormônios do crescimento. A trombopoietina é um dos principais reguladores do desenvolvimento de megacariócitos e de plaquetas, que age por meio da ligação a um receptor da superfície celular codificado pelo proto-oncogene c-mpl. Em condições normais, a regulação dos níveis plasmáticos de trombopoietina depende da produção de plaquetas.
|
*Os autores e os editores agradecem as contribuições do autor anterior, Stefan Faderl, MD, no desenvolvimento e na redação desta revisão, bem como Kate Newberry, PhD, pela assistência editorial. |
Esse feedback loop negativo falha nos casos de TE, em parte por causa da desregulação das quimiocinas e dos receptores envolvidos na megacariopoiese e trombopoiese.13 Embora o consenso geral sugira que, tipicamente, a ativação da rota de sinalização intracelular JAK-STAT seja um problema subjacente aos NMPs, há uma discussão em curso sobre o papel das mutações potenciais na patogênese da TE que ativam essa rota.
Há evidências cada vez convincentes de que a mutação genética e, possivelmente, a “dosagem genética” (isto é, a carga de alelos) estejam associadas a manifestações e resultados distintos da doença nos casos de TE e de outros NMPs. Por exemplo, características distintas foram descritas em casos de TE com mutação JAK2V617F positiva em comparação com a doença com mutação JAK2V617F negativa: contagem mais elevada de leucócitos e níveis mais altos de hemoglobina, intensificação na atividade eritroide e granulocítica na medula óssea, níveis mais baixos de eritropoietina (EPO) e taxas mais elevadas de trombose venosa.14
Alguns investigadores levantaram a hipótese de haver uma sobreposição entre TE com mutação JAK2V617F positiva e policitemia vera (PV), e que a manifestação clínica predominante depende de “modificadores” com dose genética ou da ativação de rotas distintas de sinalização intracelular. Na realidade, a grande maioria dos pacientes com TE tem cargas de alelos JAK2V617F inferiores a 50%, sugerindo que dosagens mais baixas do gene JAK2V617F possivelmente contribuam para o desenvolvimento de TE versus PV.15,16
Mais recentemente, a descoberta de mutações em CALR em, aproximadamente, 70% de pacientes com TE negativos para as mutações JAK2 ou MPL indica que poderá haver subtipos distintos de TE ou mais de uma rota genética que leve à TE. Na realidade, a mutação CALR pode definir um novo subgrupo de pacientes portadores de TE.
Em uma instituição, em uma série de pacientes com TE 717, as mutações CALR se correlacionavam com idade mais jovem, gênero masculino, baixa incidência de trombose, contagem mais elevada de plaquetas, nível mais baixo de hemoglobina e contagens mais baixas de leucócitos, em comparação com TE positiva para a mutação JAK2V617F.17 Por outro lado, a mutação JAK2V617F está associada a níveis mais elevados de hemoglobina, níveis mais baixos de plaquetas e de EPO sérica, e aumento no risco de trombose venosa.14
Em outro estudo, ao se comparar TE triplo negativa (negativa para as mutações JAK2, CALR e MPL), a mutação CALR permaneceu associada a uma contagem elevada de plaquetas e ao gênero masculino.18 Os pacientes com menor frequência da mutação MPL apresentaram nível mais baixo de hemoglobina ou contagem mais elevada de plaquetas e uma frequência maior de problemas microvasculares.19
Diagnóstico
Com frequência, as manifestações clínicas e laboratoriais da TE se sobrepõem às dos NMPs, dificultando o diagnóstico em alguns casos. A detecção de mutações em JAK2, CALR ou MPL em até 90% de pacientes com TE fez com que a distinção entre TE e outras condições reativas se tornasse mais robusta. No entanto, as distinções dentro do grupo de NMPs dependem fundamentalmente das ferramentas clínicas e laboratoriais (avaliação do sangue e da medula óssea).20 Os critérios mais recentes para o diagnóstico de TE foram revisados em 2008 pela Organização Mundial da Saúde (OMS).21
Manifestações clínicas
A apresentação clínica de pacientes com TE se caracteriza pela presença de eventos trombóticos e hemorrágicos. As complicações trombóticas se manifestam como trombose em veias profundas e embolia pulmonar. A trombose em veias hepáticas leva à síndrome de Budd-Chiari; a trombose em veias renais pode produzir a síndrome nefrótica. Eventos vasculares oclusivos na microvasculatura são responsáveis por um grande número de sintomas, incluindo eritromelalgia e isquemia digital.
As cefaleias são comuns e, em alguns casos, é muito difícil fazer a distinção entre cefaleia e enxaqueca. Os eventos hemorrágicos ocorrem em até 40% de pacientes; os sítios de sangramento incluem os tratos gastrintestinal e urinário, pele, olhos ou cérebro. Os pacientes individuais podem sofrer de episódios trombóticos e hemorrágicos.22 Cada vez mais, a TE tem sido diagnosticada em pacientes assintomáticos como descoberta incidental.
Exame Físico
O exame físico de pacientes que se apresentam com TE pode ser normal, a não ser pela presença de esplenomegalia leve observada na palpação (<5cm), que ocorre em até 40% dos casos.
Testes Laboratoriais
Alguns pacientes são assintomáticos e são diagnosticados apenas depois da descoberta de trombocitose (=450.000/µL) em rastreamentos normais feitos através de hemogramas. Outras descobertas incluem leucocitose ou, menos frequentemente, anemia leve. Tipicamente, as biópsias da medula óssea revelam a presença de hipercelularidade, variando de normal a leve, com elevação no número de megacariócitos grandes e maduros. Os testes das mutações JAK2V617F, CALR e MPLW515L são bastante úteis para excluir a hipótese de trombocitose reativa.
O Quadro 1 apresenta os critérios revisados da OMS para o diagnóstico de TE.
Quadro 1
|
CRITÉRIOS REVISADOS DA ORGANIZAÇÃO MUNDIAL DA SAÚDE PARA O DIAGNÓSTICO DE TROMBROCITEMIA ESSENCIAL |
|
1. Contagem sustentada de plaquetas =450 x 109/L.* 2. Espécime de biópsia da medula óssea mostrando proliferação, principalmente da linhagem megacariocítica, com aumento no número de megacariócitos grandes e maduros; nenhum aumento significativo no desvio esquerdo de granulopoiese ou eritropoiese neutrofílica. 3. Diagnóstico que não atenda aos critérios da OMS para PV,? MFP,? LMC,§ SMD,? ou outros neoplasmas mieloides. 4. Demonstração de JAK2V617F de outro marcador clonal, ou na ausência de um marcador clonal ou nenhuma evidência de trombocitose reativa.¶ |
LMC: leucemia mielocítica crônica; MFP: mielofibrose primária; OMS: Organização Mundial da Saúde; PV: policitemia vera; SMD: síndrome mielodisplásica.
O diagnóstico tem que atender aos quatro critérios.
*Durante o período de exame completo.
?Exige falha na terapia de reposição de ferro para elevar o nível de hemoglobina para a faixa de PV, na presença de nível sérico reduzido de ferritina. A exclusão de PV se fundamenta nos níveis de hemoglobina e de hematócrito, sendo que não é necessário medir a massa de eritrócitos.
?Exige ausência de fibrose reticulínica relevante, fibrose colagenosa, leucoeritroblastose no sangue periférico, ou medula óssea acentuadamente hipercelular para a idade acompanhada de morfologia megacariocítica típica de MFP ? com dimensões variando de pequenas a grandes, com uma proporção nuclear/citoplasmática aberrante e núcleos hipercromáticos, bulbosos ou dobrados de forma irregular e agrupamento denso.
§Exige ausência de BCR-ABL.
?Exige ausência de desieritropoiese e desgranulopoiese.
¶As causas de trombocitose reativa incluem deficiência de ferro, esplenectomia, cirurgia, infecção, inflamação, doença nos tecidos conjuntivos, câncer metastático e distúrbios linfoproliferativos. Entretanto, a presença de alguma condição associada à trombocitose reativa não excluir a possibilidade de TE se os três primeiros critérios forem atendidos.
Diagnóstico Diferencial
O diagnóstico de TE depende primeiramente da exclusão das causas secundárias de trombocitose. As causas mais frequentes de trombocitose secundária incluem deficiência de ferro, condições inflamatórias e infecções crônicas, malignidades e esplenectomia anterior. Também devem excluídos os diagnósticos de leucemia mielocítica crônica (LMC), PV, mielofibrose primária (MFP) ou síndrome mielodisplásica (SMD)/NMP.
Os testes de fusão dos genes BCR-ABL geralmente são usados para excluir a presença de LMC. A biópsia da medula óssea é imprescindível para excluir a presença de MFP e de SMD/NMP. A distinção entre TE e MFP “pré-fibrótica” também pode ser muito importante, levando-se em consideração que os pacientes com MFP pré-fibrótica correm risco maior de progressão para MFP manifesta ou de transformação para leucemia mielocítica aguda e de sobrevida mais curta.23
Ainda há muita controvérsia em torno do diagnóstico diferencial de MFP pré-fibrótica porque ela se fundamenta na identificação subjetiva de megacariócitos displásicos em biópsias na medula óssea. A despeito desse fato, a identificação de megacariócitos displásicos em pacientes com TE sugere a necessidade de monitoramento mais frequente em busca de sinais e sintomas de MF manifesta. Em 2008, o International Working Group for Myelofibrosis Research and Treatment (IWG-MRT) publicou algumas orientações para os diagnósticos pós TE-MF.24
Gerenciamento
A expectativa de vida, na grande maioria dos pacientes com TE, é normal; portanto, recomenda-se evitar tratamentos excessivos. A meta principal do tratamento é impedir a ocorrência de eventos tromboembólicos, a complicação mais fatal nos casos de TE.
De um modo geral, na ausência de contraindicações, recomendam-se baixas doses de ácido acetilsalicílico para todos os pacientes com sintomas microvasculares. A terapia citorredutora para tratamento de TE deve ser reservada para pacientes com risco elevado de complicações trombóticas (idade superior a 60 anos ou histórico de eventos trombóticos).
Esse tipo de terapia poderá ser considerado também para indivíduos com contagens de plaquetas acima de 1.500 x 109/L para diminuir o risco de hemorragias associado à deficiência no fator de von Willebrand causado por níveis elevados de plaquetas (nesse caso, deve-se evitar o uso de ácido acetilsalicílico até que o número de plaquetas esteja sob controle).
O Quadro 2 apresenta os critérios recomendados pelo International Working Group-Myelofibrosis Research and Treatment para o diagnóstico de mielofibrose pós-trombocitemia essencial.
Quadro 2
|
CRITÉRIOS RECOMENDADOS PELO INTERNATIONAL WORKING GROUP-MYELOFIBROSIS RESEARCH AND TREATMENT PARA O DIAGNÓSTICO DE MIELOFIBROSE PÓS-TROMBOCITEMIA ESSENCIAL | |
|
Exigência |
Diagnóstico anterior de TE por meio da aplicação de critérios da OMS, fibrose na medula óssea de graus 2?3 (na escala de 0?3)* ou 3?4 (na escala de 3?4)?. |
|
Exigências adicionais (são necessárias duas) |
Anemia e queda de =2mg/mL no nível de hemoglobina em relação à linha de base; leucoeritroblastose; aumento de =5cm na esplenomegalia palpável ou surgimento de esplenomegalia de palpação recente; elevação no nível de LDH; desenvolvimento de um ou mais entre os seguintes fatores: perda de peso >10% em 6 meses, sudorese noturna, febre inexplicável (>37,5ºC [99,5ºF]). |
LDH: lactato desidrogenase; OMS: Organização Mundial da Saúde; TE: trombocitemia essencial.
*Classificação europeia de fibrose na medula óssea.
?Classificação padrão de fibrose na medula óssea.
Em pacientes com risco baixo de TE e com outros fatores de risco para trombose (por exemplo, fatores de risco cardiovascular), recomenda-se envidar todos os esforços para corrigir e controlar esses fatores antes de iniciar a terapia citorredutora (alguns especialistas se referem a essa população de indivíduos como grupo de pacientes de risco intermediário).
De um modo geral, no caso de pacientes de alto risco de TE, o risco de trombose poderá ser reduzido de forma segura e eficiente com a combinação de baixas doses de ácido acetilsalicílico e terapia citorredutora. Um estudo realizado em 2005 comparou a eficácia do ácido acetilsalicílico em combinação com hidroxiureia ou anagrelida em 809 pacientes com TE com alto risco de eventos vasculares.25
O controle da contagem de plaquetas no longo prazo obteve bons resultados em ambos os grupos. Entretanto, em comparação com o grupo de hidroxiureia, os pacientes que foram tratados com anagrelida apresentaram uma probabilidade maior de ocorrência das seguintes condições: trombose arterial, hemorragia séria e transformação da MF pós-TE.
Por outro lado, a taxa de incidência de tromboembolismo venoso foi mais baixa no grupo de anagrelida. Os pesquisadores chegaram à conclusão de que as terapias à base de hidroxiureia e baixas doses de aspirina foram mais eficazes que a terapia com anagrelida.
No entanto, um estudo randomizado mais recente, realizado após a adoção dos critérios diagnósticos da OMS em 2008, demonstrou que a anagrelida não é inferior à hidroxiureia no que diz respeito à prevenção de eventos tromboembólicos e hemorrágicos em pacientes com “TE autêntica”.26 Consequentemente, embora a hidroxiureia ainda seja a terapia padrão de linha de frente para TE de alto risco, a anagrelida também é uma opção de tratamento a ser levada em consideração.
TE: trombocitemia essencial.

Figura 1 - Algoritmo de tratamento de TE.
O uso de interferon a convencional demonstrou alguma atividade na redução da contagem de plaquetas nos casos de TE, embora a toxicidade desse agente tenha sido responsável por taxas elevadas de descontinuação precoce.27 Um estudo recente mostrou que o interferon a peguilado foi mais bem tolerado que interferon convencional, além de ter reduzido a carga de alelos em 38% de pacientes com TE positivos para a mutação JAK2V617F.28
Um relatório recente que abordou dois casos de TE com CALR positiva demonstrou que houve redução na carga de alelos CALR após o tratamento com interferon a peguilado.29 A despeito do potencial para reduzir clones malignos, e considerando-se as limitações das evidências atuais e as toxicidades conhecidas, o uso de interferon a peguilado deve ser reservado como terapia de segunda linha para aplicação em pacientes intolerantes ou resistentes à terapia à base de hidroxiureia/anagrelida ou em mulheres grávidas ou com grande potencial de engravidamento.
Prognóstico
Eventos hemorrágicos e trombóticos são as complicações mais frequentes associadas à TE. Ainda não foi estabelecida uma correlação entre o grau de trombocitose e o risco de trombose.30 A avaliação padrão do risco de incidência de trombose nos casos de TE se baseia em apenas dois fatores: idade acima de 60 anos e histórico de evento trombótico.
Em 2012, o grupo IWG-MRT propôs um novo modelo prognóstico para aprimorar a previsão de risco de trombose nos casos de TE, denominado International Prognostic Score of Thrombosis in Essential Thrombocythemia (IPSET-thrombosis).31 Esse estudo utilizou os dados obtidos em 891 pacientes com TE definida pela OMS e que haviam sido tratados em sete centros internacionais.
A pontuação prognóstica final incluiu quatro fatores de risco com pesos derivados das razões de risco correspondentes na análise multivariada: idade igual ou superior a 60 anos (1 ponto); fatores de risco cardiovascular, tais como diabetes melito, hipertensão ou tabagismo (1 ponto); trombose anterior (2 pontos); e mutação JAK2V617F (2 pontos).
Usando a ponderação desses quatro fatores de risco, os pacientes foram classificados em três categorias de risco como segue: risco baixo (0 a 1 ponto), risco intermediário (2 pontos) e risco elevado (=3 pontos). A taxa anual de incidência de trombose variou de 1,03% de pacientes por ano na categoria de risco baixo a 3,56% de pacientes por ano na categoria de risco elevado.
O Quadro 3 apresenta os fatores prognósticos no IPSET e no IPSET-thrombosis.
Quadro 3
|
FATORES PROGNÓSTICOS NO INTERNATIONAL PROGNOSTIC SCORE OF THROMBOSIS IN ESSENTIAL THROMBOCYTHEMIA E NO IPSET-THROMBOSIS | ||
|
Fator de risco |
IPSET |
IPSET Thrombosis |
|
Idade =60 anos |
2 |
1
|
|
Trombose anterior |
1 |
2
|
|
Contagem de leucócitos =11 x 109/L |
1 |
-
|
|
Fatores de risco cardiovascular |
- |
1
|
|
JAK2V617F positiva |
- |
2
|
|
Pontuação prognóstica (sobrevida média ou taxa de incidência de trombose) | ||
|
Baixa |
0 pontos (não atingida) |
<2 pontos (1,03% pacientes/ano)
|
|
Intermediária |
1?2 pontos (24,5 anos) |
2 pontos (2,35% pacientes/ano)
|
|
Alta |
3?4 pontos (14,7 anos) |
< 2 pontos (3,56% pacientes/ano)
|
Subsequentemente, a mesma coorte de pacientes foi utilizada no desenvolvimento de uma pontuação prognóstica para prever a sobrevida total no momento do diagnóstico (IPSET).32 Diferentemente do IPSET-thrombosis, essa pontuação prognóstica incluía idade de 60 anos ou mais (2 pontos), contagem de leucócitos igual ou superior a 11 x 109/L (1 ponto) e trombose anterior (1 ponto) como fatores de risco independentes para a sobrevida.
Os pacientes foram, então, novamente classificados em três categorias de risco, com sobrevida mediana ainda não atingida no grupo de risco baixo e 14,7 anos para o grupo de risco alto. Ao contrário do IPSET-thrombosis, os fatores de risco cardiovascular e a mutação JAK2V617F não eram preditores da sobrevida.
O IPSET e o IPSET-thrombosis ainda precisam ser testados em estudos prospectivos para que sua utilidade seja determinada na prática clínica. A despeito desse fato, esses dados sugerem que os pacientes mais velhos com fatores de risco cardiovascular, mutação JAK2V617F ou contagens elevadas de leucócitos devem ser monitorados mais de perto.
Mielofibrose
A MFP é um distúrbio de células-tronco hematopoiéticas que se caracteriza pela presença de hematopoiese extramedular, esplenomegalia, um quadro de sangue leucoeritroblástico e graus variados de fibrose medular com hiperplasia megacariocítica acentuada e atipia. A mielofibrose (MF) pode ocorrer de novo (MFP) ou poderá evoluir a partir de PV (MF pós-PV) ou de TE (pós-TE).
Epidemiologia
A incidência de MFP varia entre 0,2 e 1,0 a cada 100 mil pessoas.1,33 Tipicamente, a MFP desenvolve-se em pessoas com idade acima de 60 anos, sendo que a idade mediana no momento do diagnóstico é de 65 anos. Todavia, ela não é uma doença exclusiva de pacientes mais velhos; cerca de 20% de pacientes com MFP têm idade inferior a 60 anos
Etiologia/Genética
A causa de MFP é desconhecida; no entanto, algumas evidências indicam que a exposição a produtos petroquímicos ou a níveis elevados de radiação podem aumentar o risco de desenvolvimento de incidência da doença.34,35
Na maior parte dos casos, a MFP não é hereditária, embora algumas pessoas possam apresentar predisposição genética para desenvolver esse tipo de doença. Por exemplo, observou-se que o haplótico JAK2 46/1 pode predispor os pacientes ao desenvolvimento de NMPs positivos para a mutação JAK2V617F.3 Um estudo de grande porte realizado na Suécia chegou à conclusão de que os parentes de primeiro grau de pacientes com MFP têm risco maior para desenvolver a doença, sugerindo que há uma predisposição genética.2
Anormalidades citogenéticas são encontradas em cerca da metade de pacientes com MFP. Embora não tenha sido descrita nenhuma alteração específica para a MFP, as anormalidades mais comuns são del(13q), del(20q), trissomia do 8 ou trissomia do 9 e anormalidades no cromossomo 1 [trissomia parcial e der(t)t(1;6)].36
Deleções únicas de 13q ou 20q ou trissomia do 9, isoladamente ou com outra anormalidade, são consideradas “favoráveis”, levando-se em consideração que estão associadas a tempos de sobrevida que se assemelham aos de pacientes com cariótipo diploide.
Anormalidades no cromossomo 5 ou 7 ou a presença de mais de três anormalidades cromossômicas (cariótipo complexo) definem cariótipos “desfavoráveis” e estão associadas a um tempo de vida total mais curto. As anormalidades no cromossomo 17 parecem estar relacionadas a um tempo menor de sobrevida.37
Patogênese
A MF é uma doença clonal estimulada por células progenitoras neoplásicas derivadas de mieloides. Esse tipo de doença se caracteriza pela presença de hematopoiese extramedular, anemia progressiva, fibrose colagenosa na medula óssea associada à angiogênese, e osteoesclerose. As células progenitoras hematopoiéticas clonais que se tornarem hipersensíveis aos efeitos de diversas citocinas, ou que tiverem adquirido mecanismos de crescimento independentes das citocinas, podem ser os pilares da etiologia de MFP.
A suprarregulação da rota JAK-STAT por meio de mutações em JAK2, MPL e CALR e outros genes potenciais contribuem parcialmente para a patogênese da MFP.38 As mutações JAK2V617F ocorrem em, aproximadamente, 60% de pacientes, sendo que a ativação nas mutações de MPLW515L é detectada em 5 a 10% de casos. As mutações CALR foram detectadas em 25 a 35% de pacientes e, aparentemente, são mutuamente exclusivas com JAK2V617F e MPLW515L.39
Embora as contribuições exatas dessas mutações para a patogênese da doença não sejam claras, estudos recentes sugerem que sua heterogeneidade pode ser subjacente à do fenótipo clínico.40 Um estudo recente envolvendo 428 pacientes com MFP demonstrou que, em comparação com a MFP positiva para a mutação JAK2V617F, a MFP positiva para CALR está associada a idades mais jovens, contagens mais baixas de leucócitos e contagens mais elevadas de plaquetas, enquanto que a MFP positiva para MPLW515L/K está associada a idades mais jovens e contagens mais baixas de leucócitos.41
A exemplo do que ocorre na TE, a mutação CALR pode melhorar o prognóstico de pacientes com MFP (sobrevida mediana de 15,9 anos), ao passo que os pacientes sem qualquer uma dessas mutações (triplo negativo) apresentam os piores prognósticos (sobrevida total mediana de 2,3 anos).
Além das três mutações deflagradoras, foram encontradas diversas outras mutações em casos de MFP, embora em frequências bem mais baixas que as mutações JAK2 e CALR. Essas menos frequentes não são mutuamente exclusivas e poderão estar presentes juntamente com outras deflagradoras. Muitas dessas mutações se localizam em genes envolvidos no controle epigenético da função celular e que não se envolvem na rota JAK-STAT (por exemplo, mutações em ASXL1, EZH2, SRSF2, CBL, IDH1/IDH2, TP53, TET2 e DNMT3).
A despeito da frequência relativamente baixa, as mutações em ASXL1, SRSF2 e EZH2 estão associadas, de forma independente, à redução no tempo de sobrevida e ASXL1, SRSF2 e IDH1/2 a um aumento no risco de transformação em leucemia aguda.42
Em particular, as mutações em ASXL1, que são encontradas em até 22% de pacientes com MFP, aparentemente são as mais lesivas e poderão ser úteis para futuras estratificações de pacientes classificados como de baixo risco pelos critérios padrões. Entretanto, os eventuais estudos prospectivos futuros deverão determinar qual é a melhor forma de fornecer informações mutacionais na prática diária.
Diagnóstico
Os critérios diagnósticos para MFP foram revisados em 2008 pela OMS.21
Manifestações Clínicas
Os pacientes com MFP poderão se apresentar com alguns sintomas ou sem sintomas nos estágios iniciais da doença ou, na medida em que a doença progride, com sintomas e sinais graves como fadiga, anemia dependente de transfusões e esplenomegalia sintomática. Condições como leucopenia e trombocitopenia também são ocorrências comuns durante o progresso da doença; no entanto, logo no início do curso da doença, alguns pacientes podem manifestar leucocitose ou trombocitose.
O Quadro 4 apresenta os critérios revisados da OMS para o diagnóstico de mielofibrose primária.
Quadro 4
|
CRITÉRIOS REVISADOS DA ORGANIZAÇÃO MUNDIAL DA SAÚDE PARA O DIAGNÓSTICO DE MIELOFIBROSE PRIMÁRIA
|
|
Critérios mais importantes 1. Presença de proliferação megacariocítica e atipia,* normalmente acompanhadas de fibrose reticulínica e/ou colagenosa ou, na ausência de fibrose reticulínica, as alterações megacariocíticas devem ser acompanhadas de um aumento na celularidade da medula óssea que se caracteriza pela proliferação granulocítica e, com frequência, por redução na eritropoiese (i.e., doença pré-fibrótica na fase celular). 2. Não atende aos critérios da OMS para PV,? LMC,? SMD,§ ou qualquer outro neoplasma mieloide. 3. Demonstração de JAK2V617F ou de qualquer outro marcador clonal (p.ex., MPLW515L/K), ou na ausência de um marcador clonal, nenhuma evidência de fibrose na medula óssea atribuível a doenças inflamatórias subjacentes ou outras doenças neoplásicas.?
Critérios menos importantes¶ 1. Leucoeritroblastose 2. Elevação no nível sérico da lactato desidrogenase 3. Anemia 4. Esplenomegalia palpável |
LMC: leucemia mielocítica crônica; OMS: Organização Mundial da Saúde PV: policitemia vera; SMD: síndrome mielodisplásica.
O diagnóstico exige o atendimento de todos os critérios mais importantes e de dois critérios menos importantes.
*Megacariócitos variando de pequenos a grandes com uma razão nuclear/citoplasmática aberrante; núcleos bulbosos hipercromáticos, bulbosos ou dobrados de forma irregular; e agrupamento denso.
? Exige falha na terapia de reposição de ferro para aumentar o nível de hemoglobina para a faixa de PV na presença de redução no nível sérico de ferritina. A exclusão de PV se fundamenta nos níveis de hemoglobina e de hematócrito, não sendo necessária a medição da massa de eritrócitos.
? Exige a ausência de BCR-ABL.
§ Exige a ausência de diseritropoiese e desgranulopoiese.
? Secundária a infecção, distúrbio autoimune ou outra condição inflamatória crônica, leucemia de células pilosas ou outro neoplasma linfoide, malignidade metastática ou mielopatias tóxicas (crônicas). Cabe observar que os pacientes portadores de condições associadas à MF reativa não são imunes à MFP, sendo que o diagnóstico deve ser considerado nesses casos se não forem atendidos outros critérios.
¶ O grau de anormalidade deve ser limítrofe ou acentuado.
Aproximadamente, a metade dos pacientes com MFP se apresenta com anemia. Sintomas como fraqueza, fadiga ou dispneia, em geral, são causados por anemia. Problemas hemorrágicos e sintomas constitucionais inespecíficos podem acompanhar o curso clínico. É muito comum a presença de sintomas constitucionais debilitantes que podem agravar substancialmente a qualidade de vida.
A fadiga é o sintoma mais comum: 84% dos pacientes se queixaram de fadiga em uma pesquisa feita na Internet.43 Outros sintomas comuns incluem sudorese noturna (56% dos pacientes), prurido (50%), dor nos ossos (47%), perda de peso não intencional (20%) e febre (18%).43
Exame físico
A esplenomegalia ocorre em até 80% de pacientes; ela pode ser maciça e reflete, pelo menos em parte, o grau de hematopoiese extramedular. Essa hematopoiese extramedular poderá levar também a uma hepatomegalia acentuada (em até 40% de pacientes), hipertensão pulmonar, asceíte, tamponamento pericárdico e compressão medular.
A esplenomegalia pode produzir desconforto abdominal, saciedade precoce e dor sob as costelas no lado esquerdo. Condições como hipertensão portal, hemorragia vertical e infartos esplênicos também podem complicar a hepatoesplenomegalia.
Exames laboratoriais
De um modo geral, os esfregaços de sangue periférico mostram a presença de leucoeritroblastose e de células em forma de gotas (mielotísica), plaquetas de grandes dimensões e mieloblastos raros. Com frequência, a aspiração na medula óssea resulta no que se conhece por “punção seca”, enquanto que a biópsia mostra um agrupamento de megacariócitos hiperplásicos de diversas dimensões, fibrose extensiva, hiperplasia megacariocítica e, eventualmente, osteoesclerose.
Outras descobertas laboratoriais incluem nível sérico elevado de lactato desidrogenase, nível elevado de proteínas de ligação com a vitamina B12 e, às vezes, hiperuricemia.
Diagnóstico Diferencial
Embora a combinação de esplenomegalia, leucoeritroblastose, anemia, má qualidade de vida e hiperplasia megacariocítica na medula óssea seja uma excelente sugestão de MFP, a MF medular propriamente dita não é específica para o diagnóstico de MFP. Além disso, foram observados diversos graus de fibrose em outros NMPs.
A SMD com fibrose deve ser excluída. A MF se caracteriza pela presença de fatores como aumento nos elementos medulares com células imaturas associados à pancitopenia e de sintomas constitucionais graves. As alterações medulares fibróticas, secundárias a outros tipos de neoplasma (por exemplo, leucemia de células pilosas, câncer metastático da próstata ou de mama) ou infecções, são considerações extremamente importantes.
O Quadro 5 apresenta os sistemas de pontuação prognóstica para mielofibrose.
Quadro 5
|
SISTEMAS DE PONTUAÇÃO PROGNÓSTICA PARA MIELOFIBROSE | |||
|
Fator |
IPSS |
D-IPSS |
D-IPSS-Plus |
|
Idade >60 anos |
1 |
1 |
1
|
|
Sintomas constitucionais |
1 |
1 |
1
|
|
Hemoglobina <10g/dL |
1 |
2 |
1
|
|
Leucócitos <25 x 109/L |
1 |
1 |
1
|
|
Blastos sanguíneos =1% |
1 |
1 |
1
|
|
Contagem de plaquetas <100 x 109/L
|
- |
- |
1
|
|
Dependência de transfusão |
- |
- |
1
|
|
Cariótipo desfavorável |
- |
- |
1
|
|
Estratificação de risco (sobrevida mediana) | |||
|
Baixo |
0 ponto (11,2 anos) |
0 ponto (não atingido)
|
0 ponto (15,4 anos) |
|
Intermediário-1 |
1 ponto (7,9 anos) |
1?2 pontos (14,2 anos) |
1 ponto (6,5 anos)
|
|
Intermediário-2 |
2 pontos (4 anos) |
3?4 pontos (4 anos) |
2?3 pontos (2,9 anos)
|
|
Alto |
=3 pontos (2,3 anos) |
5?6 pontos (1,5 anos) |
=4 pontos (1,3 anos) |
D-IPSS: Dynamic International Prognostic Scoring System; IPSS: International Prognostic Scoring System.
Prognóstico
Condições com complicações trombo-hemorrágicas, infecções e progressão para leucemia aguda estão entre os eventos fatais mais frequentes em pacientes com MF. A transformação leucêmica foi documentada em até 20 a 30% de pacientes nos primeiros 10 anos após o diagnóstico.44
Entretanto, essa estimativa de risco deriva de estudos nos quais metade dos pacientes havia sido exposta a agentes alquilantes ou à radioterapia (modalidades de tratamento que raramente são usadas na apresentação por causa das propriedades leucemogênicas conhecidas); consequentemente, a taxa de transformação leucêmica não refletia o curso natural da doença em todos os pacientes.
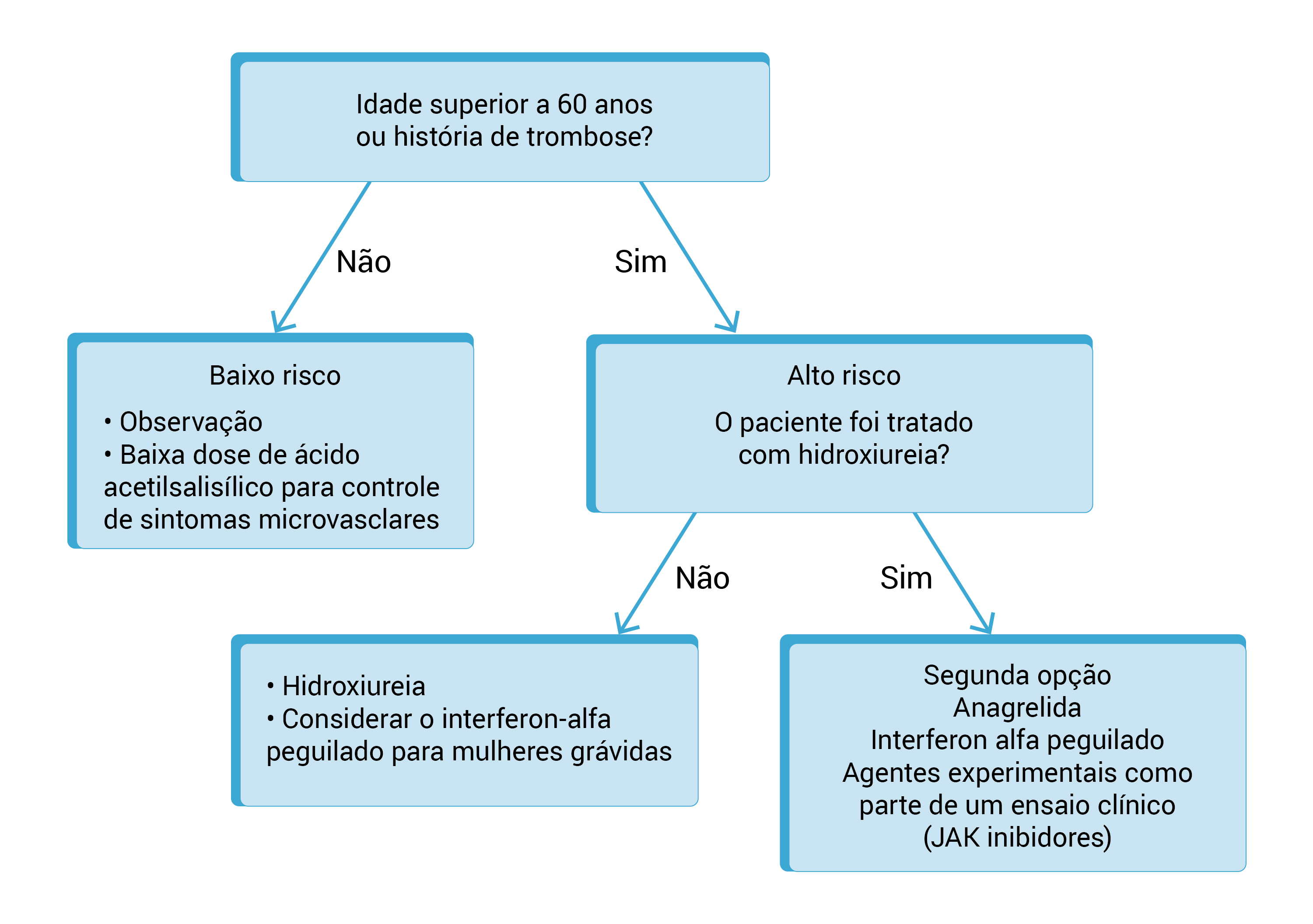
O International Prognostic Scoring System (IPSS), concebido pelo grupo IWG-MRT em 2009, é o sistema de pontuação prognóstica mais amplamente utilizado em pacientes com MFP no momento do diagnóstico. Os fatores de risco incluídos no modelo são idade acima de 65 anos, presença de sintomas constitucionais, hemoglobina inferior a 10g/dL, contagens de leucócitos acima de 25 x 109/L e blastos em circulação iguais ou superiores a 1%.
O modelo prognóstico estratifica os pacientes em quatro categorias de risco: risco baixo (0 ponto; sobrevida mediana de 11,3 anos); risco intermediário-1 (1 ponto; sobrevida mediana de 7,9 anos); risco intermediário-2 (2 pontos; sobrevida mediana de 4 anos); e risco alto (3 pontos ou mais; sobrevida mediana de 2,3 anos).
O Dynamic International Prognostic Scoring System (D-IPSS), uma versão atualizada do IPSS, trata dos fatores de risco com covariantes dependentes do tempo no modelo multivariado e, portanto, poderá ser usado para definir o prognóstico em qualquer ponto no curso do tratamento dos pacientes.
Embora os fatores de cada modelo sejam os mesmos, o D-IPSS pondera a anemia mais enfaticamente (2 pontos versus 1 ponto), o que resulta em diferenças na estratificação do risco total. O tempo de sobrevida dos pacientes estratificados nos termos do D-IPSS varia de 1,5 anos, na categoria de risco alto, a “não atingida” para os pacientes classificados como de risco baixo.
Um terceiro modelo prognóstico foi desenvolvido, o D-IPSS-plus, que inclui fatores de risco adicionais como dependência de transfusão de eritrócitos, contagem de plaquetas inferior a 100 x 109/L e cariótipo desfavorável. Esse modelo estratifica também os pacientes em quatro categorias de risco, cuja sobrevida varia de 1,3 anos (risco elevado) a 15,4 anos (risco baixo).
Mais recentemente, surgiram relatos de alguns estudos que avaliaram a relevância prognóstica de várias mutações e, provavelmente, nos próximos anos ocorrerão modificações nesses modelos prognósticos amplamente utilizados que incluirão informações mutacionais. Entretanto, ainda é necessário investigar a implementação de novas ferramentas prognósticas que incluam informações mutacionais em ambientes clínicos.
Gerenciamento
Antes da descoberta, em 2005, do papel essencial desempenhado pela sinalização JAK-STAT desregulada nos casos de MF, os tratamentos da doença eram basicamente paliativos, sua eficácia era limitada para diminuir as manifestações clínicas e, com frequência, não eram bem toleradas. As opções de tratamento são apresentadas a seguir.
Farmacoterapia Convencional
Antes da aprovação, em 2011, do medicamento ruxolitinib pela Food and Drug Administration (FDA) para tratamento de MF, as medicações citorredutoras eram usadas para controlar a hiperproliferação. Entretanto, o curso natural da doença permanecia inalterado: a hidroxiureia raramente induz a regressão completa no baço e, em raras situações, as respostas têm longa duração.
Além disso, geralmente são necessárias doses elevadas para a obtenção de respostas significativas, que poderão resultar em citopenias intoleráveis. Embora também tenham sido usados, os agentes alquilantes orais, em geral, resultam em citopenias graves e prolongadas e foram associados a um aumento no risco de transformação em leucemia aguda.
Os agentes imunomoduladores, como a talidomida ou a lenalidomida, podem reduzir a esplenomegalia e melhorar a anemia. Em um estudo, a lenalidomida em combinação com prednisona produziu respostas gerais de 30% para anemia e de 42% para esplenomegalia.45
Além disso, todos os oito pacientes com respostas positivas para a mutação JAK2V617F apresentaram uma redução na carga de alelos mutantes. As respostas foram duradouras na maioria dos pacientes. Um estudo de acompanhamento de longo prazo de pacientes que haviam sido tratados com baixas doses de talidomida mais prednisona mostrou que ocorreram melhoras semelhantes na anemia (resposta geral à anemia de 22%).46
A Figura 2 mostra o algoritmo do tratamento de MF.
EPO: eritropoietina; IPSS: International Prognostic Scoring System; MF: mielofibrose; TACT: transplante alogênico de células-tronco.
Figura 2 - Algoritmo do tratamento de MF.
Interferon a
O interferon a convencional chegou a ser avaliado como terapia para o tratamento de MF, porém as taxas de respostas foram baixas e sua toxicidade significativa impede o uso em muitos pacientes.47 Entretanto, as experiências em pacientes com MF em estado inicial sugerem que esse medicamento poderá lentificar a progressão da doença, controlar a esplenomegalia e reverter a fibrose na medula óssea em alguns pacientes.48
Entretanto, essas observações não chegaram a ser avaliadas em testes clínicos prospectivos. Mais recentemente, comprovou-se que o interferon a peguilado reduz os sintomas constitucionais e melhora a anemia, principalmente nos casos de sintomas com características proliferativas (contagem sanguínea elevada) sem esplenomegalia.49
Inibidores da JAK
O desenvolvimento dos inibidores da quinase de Janus e a aprovação recente do ruxolitinib para uso nos EUA, na Europa e no Canadá modificaram de forma significativa o panorama do tratamento de MF. A aprovação reguladora do ruxolitinib para tratamento de MF se fundamentou nos resultados de dois testes importantes de fase III, sendo que um deles comparou o ruxolitinib versus placebo (Controlled Myelofibrosis Study With Oral JAK Inhibitor Treatment [COMFORT]-I)50 e o outro fez uma comparação entre o ruxolitinib e a melhor terapia disponível (COMFORT-II).51
Em ambos os estudos, um grupo mais significativo de pacientes nos braços do ruxolitinib apresentou uma redução igual ou superior a 35% no volume do baço (aproximadamente, 50% de redução por meio de palpação) na semana 24 em comparação com a linha de base (COMFORT-I) ou na semana 48 (COMFORT-II).
O uso de ruxolitinib melhorou também os sintomas relacionados à MF e a qualidade de vida de forma mais significativa que o placebo ou a melhor terapia disponível. Embora não tenha sido comprovado que o ruxolitinib elimine o clone maligno, análises de acompanhamento de longo prazo demonstraram que os efeitos do medicamento têm longa duração e melhoram a sobrevida.52,53
Além disso, os resultados iniciais sugerem que em alguns pacientes o tratamento de longo prazo com ruxolitinib poderá retardar a progressão ou reverter a fibrose na medula óssea.54 Condições como trombocitopenia e anemia são as toxicidades mais comuns associadas ao uso do medicamento. A maior parte das toxicidades surge nas primeiras 3 semanas de tratamento e, de um modo geral, é gerenciada com reduções na dose.
A maioria dos pacientes com esplenomegalia sintomática ou com sintomas sistêmicos relacionados à MF, mesmo aqueles com anemia que depende de transfusões, pode ser tratada com sucesso com ruxolitinib por longos períodos de tempo com monitoramento cuidadoso (sobretudo durante os 3 primeiros meses), com ajuste na dose para evitar interrupções na terapia.
A recomendação atual é uma dose inicial de 20mg, 2x/dia, em pacientes com contagens de plaquetas acima de 200 x 109/L, 15mg, 2x/dia, em pacientes com contagens de plaquetas entre 100 e 200 x 109/L, 5mg, 2x/dia, em pacientes com contagens de plaquetas abaixo de 100 x 109/L. A dose poderá ser modificada de acordo com a tolerância dos pacientes até a dose máxima de 25mg, 2x/dia.
É extremamente importante evitar a interrupção do tratamento para que seja possível atingir o sucesso total, tendo em vista que os sintomas retornam à linha de base dentro de 7 a 10 dias. O grau de redução no volume do baço se correlaciona diretamente com a sobrevida de pacientes tratados com ruxolitinib.
Doses de 10mg, 2x, ou doses mais elevadas são apropriadas como doses de manutenção. Outros inibidores da JAK que são menos mielossupressivos (pacritinib) e que podem diminuir a necessidade de transfusões (momelotinib) encontram-se na fase final de desenvolvimento clínico para uso nos tratamentos de MF.
O Quadro 6 mostra a atividade clínica dos inibidores da JAK2.
Quadro 6
|
ATIVIDADE CLÍNICA DOS INIBIDORES DA JAK2 | |||
|
Medicamento |
Alvo |
Efeitos clínicos |
Estágio de desenvolvimento |
|
Ruxolitinib |
JAK1, JAK2 |
Redução na esplenomegalia e melhora no peso, no estado de desempenho e na qualidade de vida; redução nos níveis de citocina inflamatória; mielossupressão.
|
Aprovado pela FDA e pela EMA em 2011. |
|
Momelotinib |
JAK2 |
Redução na esplenomegalia; melhora na dependência de transfusões; neuropatia periférica leve; trombocitopenia.
|
Teste randomizado controlado de fase III. |
|
Pacritinib |
JAK2, FLT3 |
Redução na esplenomegalia, nenhuma mielossupressão significativa; efeitos colaterais gastrintestinais. |
Teste randomizado controlado de fase III. |
EMA: European Medicines Agency; FDA: Food and Drug Administration.
Esplenectomia e radiação esplênica
A esplenectomia está associada a níveis significativos de morbidade e mortalidade. Até 50% dos pacientes têm complicações, sendo que a mortalidade perioperatória varia de 5 a 10%.55 Um estudo mostrou que há um risco maior de transformação de blastos em pacientes que tenham se submetido a uma esplenectomia. Outra complicação é a presença de hepatomegalia progressiva e maciça como resultado de metaplasia mieloide compensatória, que poderá produzir insuficiência hepática e levar os pacientes à morte.56
Portanto, a esplenectomia é indicada para uso em pacientes com hipertensão portal sintomática, esplenomegalia grave associada à dor ou caquexia grave refratária à farmacoterapia, ou transfusões frequentes de eritrócitos e citopenias graves que não respondem aos medicamentos.
A radiação poderá produzir alívio sintomático transitório nos casos de hepatoesplenomegalia dolorida (respostas com duração mediana de 3 a 6 meses); entretanto, a radiação está associada a taxas de mortalidade acima de 10% por causa das complicações.57 Portanto, recomenda-se o uso de radiação esplênica apenas como último recurso para paliação em pacientes que não tenham respondido a outras terapias e que não sejam candidatos para esplenectomia.
Transplante Alogênico de Células-Tronco
O transplante alogênico de células-tronco (TACT) ainda é o único tratamento curativo para MF. Existem relatos de remissões duráveis em pacientes selecionados, incluindo alguns com transformação leucêmica, depois de quimioterapia de indução bem-sucedida.58 Entretanto, menos de 10% de pacientes com MF se submetem a esse tipo de procedimento por causa de fatores como idade avançada, comorbidades graves ou falta de doador.
Os pacientes com doença de risco intermediário ou de alto risco somente devem ser candidatos para TACT se apresentarem bom estado de desempenho e não tiverem nenhuma comorbidade significativa. O condicionamento com intensidade reduzida é uma opção que poderá ser usada em pacientes mais velhos e em pacientes com comorbidades.59
Ainda há controvérsias sobre o uso da esplenectomia antes do TACT e, por essa razão, não é recomendada para aplicação rotineira. Mais recentemente, a aplicação da terapia com ruxolitinib foi sugerida como ponte para o TACT.60 Encontram-se em andamento alguns estudos prospectivos para avaliar a hipótese de o pré-tratamento com ruxolitinib ser uma estratégia eficaz para melhorar os resultados depois do TACT e para aumentar potencialmente o número de pacientes elegíveis para o procedimento.
Cuidados de Suporte e Outros Agentes
A anemia progressiva com dependência de transfusão ainda é um grande problema no gerenciamento de pacientes com MF. As opções de gerenciamento de anemia relacionada à MF incluem o uso de agentes estimuladores de eritroides, esteroides, androgênios ou medicamentos imunomoduladores (talidomida ou lenalidomida, com ou sem prednisona).
As injeções subcutâneas de EPO (40.000U/semana) são opções a serem aplicadas em pacientes com nível sérico baixo de EPO (<125U/L). Corticosteroides como a prednisona (0,5 a 1,0mg/kg/dia) ou androgênios, como as injeções de enantato de testosterona (400 a 600mg/semana) e danazol por via oral (200mg, 2 ou 3x/dia), também são medicamentos comprovadamente úteis.
Os medicamentos imunomoduladores como a talidomida e a lenalidomida também melhoram a anemia associada à MF. A talidomida pode ser administrada em doses baixas (50mg/dia) em combinação com a redução gradual nas doses de prednisona (apenas nos primeiros 3 meses). Um estudo de fase II que combinou lenalidomida e prednisona nos primeiros 3 meses registrou um a resposta de 30% à hemoglobina, porém apenas em pacientes sem trombocitopenia ou neutropenia na linha de base.45
|
Informações Financeiras: Srdan Verstovsek, MD, PhD, recebeu financiamento para realização de pesquisas clínicas da empresa Incyte. Hagop Kantarjiam, MD, não possui relações financeiras relevantes a declarar. Esta revisão foi feita anteriormente por Stefan Faderl, MD, cujas informações financeiras foram apresentadas por ocasião da publicação inicial. Este texto foi revisado, atualizado e reeditado pelos autores mencionados. |
1. Titmarsh GJ, Duncombe AS, McMullin MF, et al. How common are myeloproliferative neoplasms? A systematic review and meta-analysis. Am J Hematol 2014;89:581–7.
2. Landgren O, Goldin LR, Kristinsson SY, et al. Increased risks of polycythemia vera, essential thrombocythemia, and myelofibrosis among 24 577 first-degree relatives of 11 039 patients with myeloproliferative neoplasms in Sweden. Blood 2008;112:2199–204.
3. Jones AV, Chase A, Silver RT, et al. JAK2 haplotype is a major risk factor for the development of myeloproliferative neoplasms. Nat Genet 2009;41:446–9.
4. Olcaydu D, Harutyunyan A, Jager R, et al. A common JAK2 haplotype confers susceptibility to myeloproliferative neoplasms. Nat Genet 2009;41:450–4.
5. Ranjan A, Penninga E, Jelsig AM, et al. Inheritance of the chronic myeloproliferative neoplasms. A systematic review. Clin Genet 2013;83:99–107.
6. Gangat N, Tefferi A, Thanarajasingam G, et al. Cytogenetic abnormalities in essential thrombocythemia: prevalence and prognostic sig-nificance. Eur J Haematol 2009;83:17–21.
7. Baxter EJ, Scott LM, Campbell PJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005;365:1054–61.
8. Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med 2005;352:1779–90.
9. Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005;7:387–97.
10. Pikman Y, Lee BH, Mercher T, et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med 2006;3:e270.
11. Klampfl T, Gisslinger H, Harutyunyan AS, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med 2013;369:2379–90.
12. Nangalia J, Massie CE, Baxter EJ, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med 2013;369:2391–405.
13. Salim JP, Goette NP, Lev PR, et al. Dysregulation of stromal derived factor 1/CXCR4 axis in the megakaryocytic lineage in essential thrombocythemia. Br J Haematol 2009;144:69–77.
14. Campbell PJ, Scott LM, Buck G, et al. Definition of subtypes of essential thrombocythaemia and relation to polycythaemia vera based on JAK2 V617F mutation status: a prospective study. Lancet 2005;366:1945–53.
15. Antonioli E, Guglielmelli P, Poli G, et al. Influence of JAK2V617F allele burden on phenotype in essential thrombocythemia. Haematologica 2008;93:41–8.
16. Kittur J, Knudson RA, Lasho TL, et al. Clinical correlates of JAK2V617F allele burden in essential thrombocythemia. Cancer 2007;109:2279–84.
17. Rumi E, Pietra D, Ferretti V, et al. JAK2 or CALR mutation status defines subtypes of essential thrombocythemia with substantially dif-ferent clinical course and outcomes. Blood 2014;123:1544–51. DOI: 10.1182/blood-2013-11-539098. [Epub 2013 Dec 23]
18. Tefferi A, Wassie EA, Lasho TL, et al. Calreticulin mutations and long-term survival in essential thrombocythemia. Leukemia 2014;28:2300–3. DOI: 10.1038/leu.2014.148. [Epub 2014 May 5]
19. Vannucchi AM, Antonioli E, Guglielmelli P, et al. Characteristics and clinical correlates of MPL 515W>L/K mutation in essential throm-bocythemia. Blood 2008;112:844–7.
20. Wadleigh M, Tefferi A. Classification and diagnosis of myeloproliferative neoplasms according to the 2008 World Health Organization criteria. Int J Hematol 2010;91:174–9.
21. Swerdlow SH, International Agency for Research on Cancer, World Health Organization. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon (France): International Agency for Research on Cancer; 2008.
22. Michiels JJ, Berneman Z, Van Bockstaele D, et al. Clinical and laboratory features, pathobiology of platelet-mediated thrombosis and bleeding complications, and the molecular etiology of essential thrombocythemia and polycythemia vera: therapeutic implications. Semin Thromb Hemost 2006;32:174–207.
23. Barbui T, Thiele J, Passamonti F, et al. Survival and disease progression in essential thrombocythemia are significantly influenced by accurate morphologic diagnosis: an international study. J Clin Oncol 2011;29:3179–84.
24. Barosi G, Mesa RA, Thiele J, et al. Proposed criteria for the diagnosis of post-polycythemia vera and post-essential thrombocythemia myelofibrosis: a consensus statement from the International Working Group for Myelofibrosis Research and Treatment. Leukemia 2008;22:437–8.
25. Harrison CN, Campbell PJ, Buck G, et al. Hydroxyurea compared with anagrelide in high-risk essential thrombocythemia. N Engl J Med 2005;353:33–45.
26. Gisslinger H, Gotic M, Holowiecki J, et al. Anagrelide compared with hydroxyurea in WHO-classified essential thrombocythemia: the ANAHYDRET Study, a randomized controlled trial. Blood 2013;121:1720–8.
27. Samuelsson J, Hasselbalch H, Bruserud O, et al. A phase II trial of pegylated interferon alpha-2b therapy for polycythemia vera and es-sential thrombocythemia: feasibility, clinical and biologic effects, and impact on quality of life. Cancer 2006;106:2397–405.
28. Quintas-Cardama A, Kantarjian H, Manshouri T, et al. Pegylated interferon alfa-2a yields high rates of hematologic and molecular re-sponse in patients with advanced essential thrombocythemia and polycythemia vera. J Clin Oncol 2009;27:5418–24.
29. Cassinat B, Verger E, Kiladjian JJ. Interferon alfa therapy in CALR-mutated essential thrombocythemia. N Engl J Med 2014;371:188–9.
30. Schafer AI. Thrombocytosis. N Engl J Med 2004;350:1211–9.
31. Barbui T, Finazzi G, Carobbio A, et al. Development and validation of an International Prognostic Score of thrombosis in World Health Organization-essential thrombocythemia (IPSET-thrombosis). Blood 2012;120:5128–33; quiz 252.
32. Passamonti F, Thiele J, Girodon F, et al. A prognostic model to predict survival in 867 World Health Organization-defined essential thrombocythemia at diagnosis: a study by the International Working Group on Myelofibrosis Research and Treatment. Blood 2012;120:1197–201.
33. Mehta J, Wang H, Iqbal SU, Mesa R. Epidemiology of myeloproliferative neoplasms in the United States. Leuk Lymphoma 2014;55:595–600.
34. Arnold AG, Oelbaum MH. Thorotrast administration followed by myelofibrosis. Postgrad Med J 1980;56:124–7.
35. Hu H. Benzene-associated myelofibrosis. Ann Intern Med 1987;106:171–2.
36. Hussein K, Van Dyke DL, Tefferi A. Conventional cytogenetics in myelofibrosis: literature review and discussion. Eur J Haematol 2009;82:329–38.
37. Hussein K, Pardanani AD, Van Dyke DL, et al. International Prognostic Scoring System-independent cytogenetic risk categorization in primary myelofibrosis. Blood 2010;115:496–9.
38. Rampal R, Al-Shahrour F, Abdel-Wahab O, et al. Integrated genomic analysis illustrates the central role of JAK-STAT pathway activation in myeloproliferative neoplasm pathogenesis. Blood 2014;123:e123–33.
39. Rumi E, Pietra D, Pascutto C, et al. Clinical effect of driver mutations of JAK2, CALR, or MPL in primary myelofibrosis Blood 2014;124:1062–9.
40. Cazzola M, Kralovics R. From Janus kinase 2 to calreticulin: the clinically relevant genomic landscape of myeloproliferative neoplasms. Blood 2014;123:3714–9.
41. Tefferi A, Guglielmelli P, Larson DR, et al. Long-term survival and blast transformation in molecularly-annotated essential thrombo-cythemia, polycythemia vera and myelofibrosis. Blood 2014;124:2507–13; quiz 2615. DOI: 10.1182/blood-2014-05-579136. [Epub 2014 Jul 18]
42. Vannucchi AM, Lasho TL, Guglielmelli P, et al. Mutations and prognosis in primary myelofibrosis. Leukemia 2013;27:1861–9.
43. Mesa RA, Niblack J, Wadleigh M, et al. The burden of fatigue and quality of life in myeloproliferative disorders (MPDs): an international Internet-based survey of 1179 MPD patients. Cancer 2007;109:68–76.
44. Cervantes F, Tassies D, Salgado C, et al. Acute transformation in nonleukemic chronic myeloproliferative disorders: actuarial probability and main characteristics in a series of 218 patients. Acta Haematol 1991;85:124–7.
45. Quintas-Cardama A, Kantarjian HM, Manshouri T, et al. Lenalidomide plus prednisone results in durable clinical, histopathologic, and molecular responses in patients with myelofibrosis. J Clin Oncol 2009;27:4760–6.
46. Thapaliya P, Tefferi A, Pardanani A, et al. International Working Group for Myelofibrosis Research and Treatment response assessment and long-term follow-up of 50 myelofibrosis patients treated with thalidomide-prednisone based regimens. Am J Hematol 2011;86:96–8.
47. Gilbert HS. Long term treatment of myeloproliferative disease with interferon-alpha-2b: feasibility and efficacy. Cancer 1998;83:1205–13.
48. Silver RT, Vandris K, Goldman JJ. Recombinant interferon-a may retard progression of early primary myelofibrosis: a preliminary report. Blood 2011;117:6669–72. DOI: 10.1182/blood-2010-11-320069. [Epub 2011 Apr 25]
49. Ianotto JC, Boyer-Perrard F, Gyan E, et al. Efficacy and safety of pegylated-interferon alpha-2a in myelofibrosis: a study by the FIM and GEM French cooperative groups. Br J Haematol 2013;162:783–91.
50. Verstovsek S, Mesa RA, Gotlib J, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med 2012;366:799–807.
51. Harrison C, Kiladjian JJ, Al-Ali HK, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med 2012;366:787–98.
52. Cervantes F, Vannucchi A, Kiladjian J, et al. Three-year efficacy, safety, and survival from COMFORT-II, a phase 3 study comparing ruxolitinib with best available therapy for myelofibrosis. Blood 2013;122:4047–53.
53. Verstovsek S, Mesa R, Gotlib J, et al. Efficacy, safety, and survival with ruxolitinib in patients with myelofibrosis: results of a median 3-year follow-up of COMFORT-I. Haematologica 2015;100:479–88.
54. Kvasnicka HM, Thiele J, Bueso-Ramos CE, et al. Effects of five-years of ruxolitinib therapy on bone marrow morphology in patients with myelofibrosis and comparison with best available therapy. Blood 2013;122:4047–53.
55. Tefferi A, Mesa RA, Nagorney DM, et al. Splenectomy in myelofibrosis with myeloid metaplasia: a single-institution experience with 223 patients. Blood 2000;95:2226–33.
56. Mesa RA, Nagorney DS, Schwager S, et al. Palliative goals, patient selection, and perioperative platelet management: outcomes and lessons from 3 decades of splenectomy for myelofibrosis with myeloid metaplasia at the Mayo Clinic. Cancer 2006;107:361–70.
57. Mesa RA, Tefferi A. Surgical and radiotherapeutic approaches for myelofibrosis with myeloid metaplasia. Semin Oncol 2005;32:403–13.
58. Ciurea SO, de Lima M, Giralt S, et al. Allogeneic stem cell transplantation for myelofibrosis with leukemic transformation. Biol Blood Marrow Transplant 2010;16:555–9.
59. Kroger N, Holler E, Kobbe G, et al. Allogeneic stem cell transplantation after reduced-intensity conditioning in patients with myelofibrosis: a prospective, multicenter study of the Chronic Leukemia Working Party of the European Group for Blood and Marrow Transplantation. Blood 2009;114:5264–70.
60. Jaekel N, Behre G, Behning A, et al. Allogeneic hematopoietic cell transplantation for myelofibrosis in patients pretreated with the JAK1 and JAK2 inhibitor ruxolitinib. Bone Marrow Transplant 2014;49:179–84. DOI: 10.1038/bmt.2013.173. [Epub 2013 Dec 2].
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.