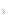 (Carregando Índice)... (Carregando Índice)... |
Autor:
Rodrigo Antonio Brandão Neto
Médico Assistente da Disciplina de Emergências Clínicas do Hospital das Clínicas da Faculdade de Medicina da USP
Última revisão: 23/08/2013
Comentários de assinantes: 0
Paciente de 74 anos de idade refere quadro de sangramentos cutâneos espontâneos há cerca de 2 meses, chega ao PS com hematomas extensos em membros inferiores e superiores e hematúria significativa. Colheu exames que revelaram alteração de TTPA com R de 6,5, não apresentava alterações em outros exames de coagulação como TP e TT e a contagem de plaquetas era normal. A Figura 1 mostra hematomas extensos que ocorreram neste paciente.
Figura 1. Hematomas extensos em membros superiores.

O diagnóstico diferencial do paciente com alteração isolada de TTPA envolve necessariamente doenças que interfiram com a via intrínseca da coagulação. Isso inclui a síndrome de anticorpos antifosfolípides, que alargam o TTPA, mas cursam com aumento do risco de trombose, e não sangramentos. Outras causas de aumento são a deficiência de Von Willebrand, que também altera a função plaquetária e o tempo de sangramento, e a coagulação intravascular disseminada (CIVD) em uma fase inicial, que eventualmente teria esta apresentação. Contudo, é comum que os pacientes tenham plaquetopenia na apresentação e alargamento de outros tempos de coagulação.
As hemofilias cursam com alteração isolada de TTPA, mas é pouco provável que o paciente só viesse a apresentar quadro de sangramento com 74 anos de idade, o que nos leva à principal possibilidade diagnóstica: a hemofilia adquirida.
Na hemofilia adquirida, ocorre a produção de anticorpos conhecidos como inibidores da coagulação, principalmente contra o fator VIII levando a aparecimento de sangramentos e alteração do TTPA.
A forma clássica de hemofilia produz uma tendência para hemorragias desde a infância, relacionada com a diminuição na concentração de fator VIII ou IX (doença de Christmas). A herança é ligada ao sexo, de forma que o sexo masculino é muito mais acometido.
Em contraste, a forma adquirida ocorre sobretudo após a 4ª década de vida e acomete ambos os sexos igualmente. Devido ao desenvolvimento de anticorpos contra o fator VIII, temos uma diminuição da concentração deste e tendência para hemorragias significativas.
O padrão das hemorragias, porém, apresenta importantes diferenças com a hemofilia clássica, sendo que as hermatroses – manifestação principal da forma clássica – são raras na forma adquirida e as principais manifestações são hemorragias cutâneas e de tecidos moles. O sangramento em tecidos moles pode piorar rapidamente e causar síndrome compartimental. Outras manifestações incluem hematúria, hemorragia gastrintestinal e hemorragia pós-parto prolongada.
Os exames laboratoriais tipicamente mostram alterações da tromboplastina parcial ativada (TTPA) e baixas concentrações de fator VIII. O tempo de trombina e protrombina é normal, assim como a contagem de plaquetas e sua função. A pesquisa dos inibidores é positiva, embora não facilmente disponível em vários centros. Os inibidores podem ser contra o fator VIII e menos comumente contra o fator IX. São anticorpos IgG-4 geralmente policlonais (IgM ou raramente IgA).
O tratamento se baseia no controle de urgência do sangramento e em reduzir a produção de autoanticorpos inibidores. Em pacientes com sangramento leve, o DDAVP em dose de 0,3 mcg/kg SC por dia, por 3 a 5 dias, pode ser suficiente. Em casos mais graves, pode-se utilizar concentrado de fator VIII em altas doses, como 20 unidades/kg para cada unidade de inibidores, com dose extra de 40 unidades/kg e monitoração da atividade do fator VIII. Em casos sem resposta, pode-se utilizar ainda o complexo protrombínico e o fator recombinante humano VIIa. Deve-se lembrar que, mesmo em altas doses do fator VIII, é provável que ele seja rapidamente inutilizado pelos inibidores presentes na circulação; assim, seu uso é limitado para os sangramentos na emergência.
A eliminação dos inibidores pode ser conseguida com a imunossupressão, podendo ser utilizados glicocorticoides isolados ou em combinação com outros imunossupressores. A dose usual de corticosteroides é de 1 mg/kg de prednisona e a associação habitual com a ciclofosfamida por via oral 2 mg/kg ou, segundo alguns autores, de 50 a 100 mg/dia. A azatioprina é um agente imunossupressor alternativo, assim como existem relatos do uso da ciclosporina também. O tratamento deve ser continuado por cerca de 2 meses, com acompanhamento periódico por especialista.
O rituximabe tem sido estudado recentemente também para este propósito, em virtude de seu sucesso em outras doenças, como a púrpura trombocitopênica idiopática. Seu uso é restrito a pacientes refratários e pode ser realizado ou não em combinação com outras terapias imunossupressoras, como ciclofosfamida e prednisona.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.