 (Carregando Índice)... (Carregando Índice)... |
Última revisão: 21/07/2014
Comentários de assinantes: 0
Uma paciente do sexo feminino, 35 anos, negra, foi admitida no hospital com sintomas de mialgias, dor óssea, febre, astenia, perda de peso e lesões pruriginosas no escalpo cobertas por crostas e com secreção seropurulenta. A paciente relatou ter apresentado pneumonia um mês antes de sua administração, infestações frequentes por parasitas intestinais, especialmente ascaridíase, e lesões no escalpo desde a infância concomitantes a várias infecções secundárias, inclusive um episódio de miíase. Ao realizar exame, foram verificadas lesões eczematoides e ceratóticas com sinais de cronicidade na região auricular, escalpo e pele adjacente; lesões crostosas escoriadas nos membros superiores e inferiores compatível com escabiose, adenomegalia na região cervical bilateral, hepatoesplenomegalia e distensão abdominal. Nos exames complementares, foi possível observar os seguintes resultados: hemoglobina de 13,7 g/dL, leucócitos totais de 380.000/µL (7% de neutrófilos, 1% de eosinófilos, 92% de linfócitos, dos quais 52% evidenciaram polimorfismo e alguns com característica de “células em flor”), contagem de plaquetas de 263.000/µL; desidrogenase lática (LDH) de 944 U/L; cálcio sérico de 9,4 mg/dL; anti-HTLV 1 positivo; medulogramahipercelular com 50% de linfócitos pleomórficos e atípicos. A partir da imunofenotipagem, constataram-se: 99% de células CD2+, CD3+, CD4+, CD5+, CD7-, CD8-, receptor de células T alfa/beta+, receptor de células T gama/delta-, CD16-, CD56-, CD57- e CD1a- em linfócitos T. As tomografias computadorizadas evidenciaram adenopatias cervicais e axilares difusas bilaterais, aumento do baço e do fígado.
As neoplasias linfoides são derivadas de células que se desenvolvem geralmente em linfócitos T (citotóxicos, helper, regulatórios) ou linfócitos B (linfócitos ou plasmócitos).
Historicamente, as neoplasias linfoides que envolvem o sangue ou a medula óssea (leucemia) têm sido classificadas separadamente daquelas que se apresentam como uma massa (linfoma). Entretanto, hoje se considera que qualquer “linfoma” pode apresentar-se como ou desenvolver um quadro leucêmico, e qualquer “leucemia” pode ocasionalmente apresentar-se com uma lesão em massa.
Na classificação da Organização Mundial da Saúde (OMS)1 (Fig. 60.1), o diagnóstico das várias neoplasias linfoides depende não da localização anatômica das células tumorais, mas da célula de origem do tumor, avaliado por morfologia, imunofenótipo e achados genéticos. Como resultado, várias entidades previamente consideradas distintas são agora agrupadas sob categorias diagnósticas únicas, como, por exemplo:
•Leucemia linfoblástica de precursores B e linfoma linfoblástico pré-B.
•Leucemia linfoblástica pré-T e linfoma linfoblásticopré-T.
•Leucemia linfocítica crônica e linfoma linfocítico de pequenas células.

Figura 60.1
Organização conceitual das neoplasias linfoides.
NK, natural killer.
Neste capítulo, são abordadas a leucemia linfoide aguda e a leucemia linfocítica crônica.
A LLA constitui um grupo de neoplasias linfoides que se assemelham às células precursoras de linhagem B ou T do ponto de vista morfológico e imunofenotípico. Essas neoplasias podem se apresentar predominantemente como um processo leucêmico, com envolvimento extensivo da medula óssea e do sangue periférico, ou podem limitar-se à infiltração tecidual, em que o envolvimento da medula óssea inexiste ou é apenas limitado (menos de 25%). Nesse último caso, designa-se linfomas linfoblásticos (LBL). A LLA e o LBL parecem constituir um contínuo biológico, embora possam mostrar características clínicas distintas. A atual classificação das neoplasias hematopoiéticas da OMS1 determina esses distúrbios como leucemia/linfoma linfoblástico B ou T.
A LLA é um distúrbio neoplásico de linfoblastos imaturos comprometido com a linhagem B ou T. Os
precursores B surgem na medula óssea, e a maioria dos pacientes com esse tumor apresentam envolvimento da medula óssea e um quadro leucêmico no sangue periférico (leucemia linfoide aguda de precursores de células B). Os precursores T podem ser originados no timo ou na medula óssea.
A maioria dos casos de LLA ocorre na infância, com uma incidência nos EUA de 3 a 4/100.000 de 0 a 14 anos, e 1/100.000 acima de 15 anos. Em crianças, a LLA corresponde a 75% dos casos de todas as leucemias agudas, as quais representam 34% de todas as neoplasias malignas nesse grupo etário, com pico de incidência de 2 a 5 anos de idade. Essa porcentagem é muito mais baixa em adultos, nos quais a leucemia mieloide aguda (LMA) e a leucemia linfocítica crônica (LLC) incidem com muito mais frequência. Há um leve predomínio da doença em indivíduos do sexo masculino em todas as idades e uma significativa incidência em crianças brancas.
43536
A LLA representa primariamente uma doença de novo, ocorrendo apenas raros casos como neoplasias secundárias. Diversos fatores genéticos e ambientais têm sido relacionados à LLA. Ela incide mais frequentemente em pacientes com síndrome de Down, síndrome de Bloom, neurofibromatose tipo I e ataxia-telangiectasia. Adicionalmente, a exposiçãoin utero à radiação ionizante, aos pesticidas e aos solventes têm sido também associada a um risco aumentado de leucemia na infância.
A maioria dos pacientes com LLA apresenta um cariótipo anormal ou no número de cromossomos (ploidia), ou mudanças estruturais, como translocações, inversões ou deleções. Entre os pacientes com alterações no número, 51% apresentam pseudodiploidia, 37%, hiperdiploidia e 12%, hipodiploidia. Entre as crianças com LLA-T, cerca de 60% evidenciam um cariótipo anormal. Pode-se observar um padrão distinto de anormalidades cariotípicas recorrentes nesses pacientes, envolvendo tanto o receptor das células T (TCR) quanto lócus não gene TCR. As alterações citogenéticas têm significado prognóstico nas LLA-B, porém não nas LLA-T.
Inicialmente, a apresentação clínica da LLA é mais frequentemente aguda, embora um pequeno percentual de casos possa desenvolver-se de forma insidiosa por vários meses. Os sintomas/sinais iniciais correlacionam-se com a massa de células leucêmicas e o grau de reposição da medula óssea, causando citopenias.
Os sintomas mais comuns são febre (causada pela leucemia ou secundária à infecção pela neutropenia), fadiga e letargia/fadiga (como resultado da anemia), dor óssea e articular e diátese hemorrágica (relacionada à trombocitopenia).
Os pacientes com precursores LLA-T frequentemente apresentam uma massa mediastinal com ou sem derrame pleural associado, que pode ocasionar disfunção respiratória e outros sinais de síndrome da veia cava superior. Sítios de envolvimento extramedular comuns incluem linfonodos, fígado, baço e meninges e, menos comumente, infiltração de órbita, testículos, tonsilas e adenoides.
As anormalidades laboratoriais evidenciadas frequentemente em casos de LLA são anemia, trombocitopenia, neutropenia e leucopenia ou leucocitose, havendo hiperleucocitose (> 100.000/ L) em cerca de 15% dos pacientes pediátricos. Outras anormalidades laboratoriais comuns são ácido úrico e desidrogenase láctica elevados, correlacionando-se à massa tumoral e ao grau de lise tumoral.
A característica principal da leucemia aguda é a combinação de pancitopenia com blastos circulantes. Entretanto, os blastos podem inexistir no sangue periférico em cerca de 10% dos casos (leucemia aleucêmica). A medula óssea é geralmente hipercelular e dominada por blastos, sendo necessários mais de 20% de blastos para o diagnóstico de leucemia aguda.
Na LLA, especialmente T, pode haver uma massa mediastinal visível no raio X de tórax. A leucemia meníngea, com blastos no líquido cerebrospinal, é observada em cerca de 5% dos pacientes ao diagnóstico.
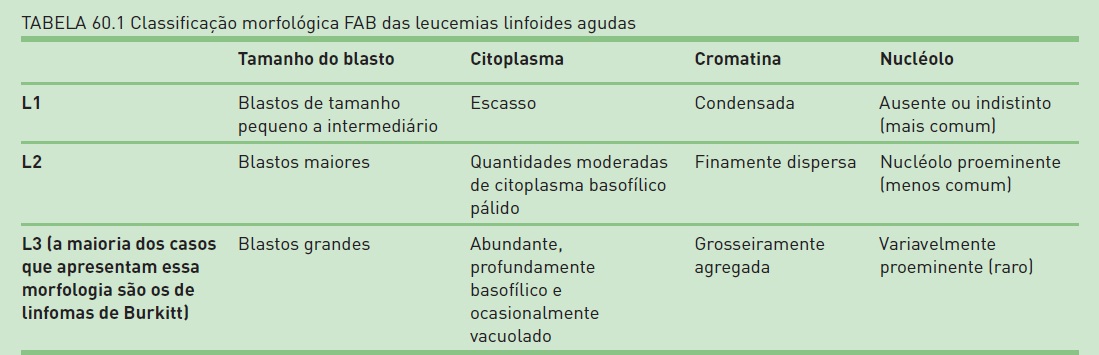
As características morfológicas clássicas da LLA são melhor resumidas pelas categorias do sistema de classificação Franco-Americano-Britânico (FAB), que se baseia na aparência microscópica das células leucêmicas e define os três tipos morfológicos: L1, L2 e L3 (Tab. 60.1).
Os blastos da LLA são frequentemente positivos para a coloração ácido periódico-Schiff (PAS) (75%) e também podem ser positivos para fosfatase ácida, mais frequentemente na LLA-T. A coloração citoquímica tem sido utilizada com menos frequência no diagnóstico da LLA devido à disponibilidade da imunofenotipagem.
A célula leucêmica retém as propriedades da linhagem de sua derivação. O fenótipo das células leucêmicas é geralmente evidenciado por meio de citometria de fluxo. As LLA são classificadas pelo imunofenótipo comoLLA-B e LLA-T. Quase todos os blastos da LLA expressam a deoxinucleotidiltransferase terminal (TdT).
Os blastos da LLA-B expressam uma variedade de antígenos específicos das células B: CD19, CD20, CD22, CD24, CD79a e, na maioria dos casos, CD10, conhecido também como antígeno comum da LLA.
Os blastos da LLA-T caracterizam-se pela expressão dos antígenos associados à linhagem T, mas, em geral, não expressam os marcadores T de superfície, como CD3, CD4 e CD8, mas alguma combinação de CD2, CD5 e CD7, e não expressam imunoglobulina de superfície.
Na LLA, algumas alterações citogenéticas são associadas a um prognóstico favorável, como a hiperdiploidia, ou desfavorável, como a translocação 9,22 (cromossomo Filadélfia) e a t(4,11).
A LLA deve ser diferenciada de outras doenças linfoproliferativas e também da linfocitose atípica que ocorre na mononucleose e na coqueluche.
Na maioria dos centros, o tratamento da LLA é realizado com quimioterapia intensiva de curto prazo (de indução), com altas doses de metotrexato, citarabina, ciclofosfamida, dexametasona, prednisona, vincristina, L-asparaginase e/ ou antraciclina. Para continuação do tratamento, realiza-se terapia de intensificação ou consolidação para eliminar leucemia residual, prevenir ou erradicar leucemia no SNC e garantir a continuidade da remissão. A radioterapia pode ser utilizada em pacientes que apresentam leucemia no SNC ou testicular, mas essa abordagem é atualmente controversa, em especial para crianças.
Em adultos, o uso de fatores de crescimento, como o fator estimulante de colônia de granulócitos que acelera a recuperação hematopoiética, tem aumentado muito a taxa de sucesso no tratamento da LLA.
Um número crescente de estudos farmacogenéticos tem observado que polimorfismos de linha germinativa e mutações na LLA podem afetar os níveis de expressão e a funcionalidade dos genes de metabolismo dos fármacos. Eles podem causar um aumento na probabilidade de leucemia em seus carreadores, influenciar a resposta dos blastos leucêmicos a agentes quimioterápicos específicos e podem também aumentar a probabilidade do desenvolvimento de malignidades relacionadas ao tratamento. Dessa forma, os regimes quimioterápicos devem ser adaptados não apenas pelas características específicas de cada leucemia aguda, mas também ao perfil genético individual do paciente.
A frequência total de recaída na LLA é de cerca de 25% na faixa pediátrica e de 50% nos adultos, dependendo muito do subtipo imunofenotípico e genético, ou seja, da categoria de risco definida. A maioria das recaídas ocorre nos primeiros três a cinco anos do diagnóstico, uma percentagem muito pequena em mais de cinco anos após o diagnóstico e, em uma minoria do pacientes, 10 a 20 anos mais tarde. A recaída pode afetar a medula óssea ou os tecidos extramedulares mais frequentemente nos sítios “santuários”, como SNC, testículos, ovários ou ambos.
O prognóstico da LLA tem melhorado consideravelmente nas últimas décadas como resultado da adaptação da terapia ao nível de risco de recaída, melhora dos cuidados de suporte e otimização dos quimioterápicos disponíveis. A sobrevida total na população pediátrica está em torno de 80 a 90%, enquanto nos adultos fica em torno de 40 a 50%.
Os pacientes com LLA são estratificados e tratados de acordo com algoritmos de estratificação de risco (Tab. 60.2). Os indicadores prognósticos mais úteis na LLA são idade, contagem de leucócitos, imunofenótipo e cariótipo.
A LLC é a leucemia linfoide de incidência mais comum e que se caracteriza por uma expansão clonal de linfócitos B maduros. Ela apresenta-se inicialmente como leucemia (LLC) ou como linfoma (linfoma linfocítico pequeno), nesse caso, representando cerca de 7% dos linfomas não Hodgkin.
A LLC é uma doença em geral indolente e caracterizada pelo acúmulo monoclonal progressivo de pequenos linfócitos funcionalmente incompetentes e com longa sobrevida. Os pacientes comumente desenvolvem complicações associadas à disfunção imune intrínseca que resulta em imunodeficiência e no desenvolvimento de distúrbios autoimunes.
437
438
A LLC é a leucemia de incidência mais frequente em países ocidentais, correspondendo a aproximadamente 30% de todas as leucemias nos EUA. Ela é uma doença de pacientes idosos, ocorrendo 90% dos casos após os 50 anos e com uma idade média de apresentação de 70 anos.
As neoplasias de linfócitos B mais comuns são originadas por células que passaram por uma reação do centro germinativo, iniciada quando linfócitos B estimulados por antígenos migram para dentro dos centros germinativos ou dos folículos dos órgãos linfoides secundários (linfonodos, baço e tecido linfoide associado à mucosa). Os linfócitos B dos centros germinativos proliferam e sofrem dois eventos que permitem a diversificação dos genes das imunoglobulinas (Ig):
•Hipermutação somática;
•Mudança da classe da cadeia pesada.
A maioria dos tumores de linfócitos B maduros apresenta hipermutação somática, e considera-se que esses erros que ocorrem durante a hipermutação somática e a mudança de classe possam ser responsáveis por muitas das mutações adquiridas que ocasionam a transformação do linfócito B. Em cerca de 60% dos casos de LLC, os genes Ig são somaticamente hipermutados; nos casos restantes, parecem derivados de linfócitos B simples.
A LLC manifesta-se clinicamente por imunossupressão, falência da medula óssea e infiltração dos órgãos com linfócitos. Muitos pacientes descobrem a doença de forma incidente devido à evidência de linfocitose em hemograma de rotina. Outros apresentam fadiga ou adenopatia. No exame, podem-se observar adenopatias em 80% dos pacientes e hepatomegalia e/ou esplenomegalia em 50%.
Em cerca de 5% dos casos de LLC, enquanto a doença permanece sistemicamente estável, um linfonodo isolado pode transformar-se em um linfoma agressivo de grandes células, caracterizando a síndrome de Richter.
O principal indício de LLC é a linfocitose isolada. O diagnóstico da LLC B típica é realizado quando há um número elevado de linfócitos circulantes, isto é, maior do que 4.000/L, e geralmente maior do que 10.000/ L, que são linfócitos B monoclonais expressando o antígeno CD5. O número de leucócitos é em geral maior do que 20.000/ L e pode ser acentuadamente elevado a muitos milhares. Entre as células circulantes, 75 a 98% são linfócitos, que parecem pequenos e maduros, morfologicamente indistintos dos pequenos linfócitos normais. O esfregaço do sangue periférico tipicamente apresenta várias células “mancha”ou “cesta”, que são restos nucleares das células danificadas pela tensão de cisalhamento ao realizar o esfregaço. Na apresentação, a hemoglobina e a contagem de plaquetas estão em geral normais.
A medula óssea é variavelmente infiltrada por pequenos linfócitos, cujo imunofenótipo evidencia coexpressão do marcador de linhagem de linfócito B CD19 com o marcador de linfócito T CD5.
Se a apresentação primária é a linfadenopatia e uma biópsia de linfonodo é realizada, os patologistas geralmente têm um pouco de dificuldade em realizar o diagnóstico de linfoma linfocítico pequeno com base nos achados morfológicos e imunofenotípicos. Entretanto, mesmo nesses pacientes, 70 a 75% apresentam envolvimento da medula óssea, e linfócitos B monoclonais circulantes estão frequentemente presentes.
A hipogamaglobulinemia ocorre em 50% dos pacientes e torna-se mais comum quando a doença está em estágio avançado. Em alguns pacientes, há uma pequena quan- tidade de paraproteína IgM.
A avaliação do paciente deve incluir um hemograma completo, bioquímica para avaliar a função dos órgãos, eletroforese de proteínas séricas e uma biópsia de medula óssea. Frequentemente são realizados estudos de imagem do tórax e do abdome para avaliar linfadenopatia patológica.
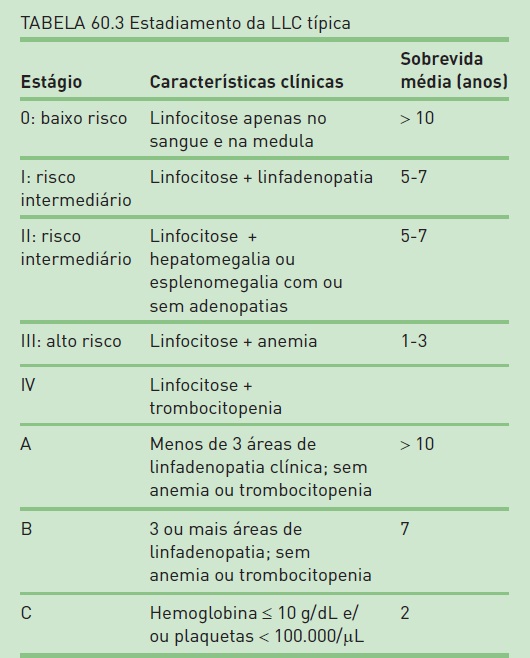
Os pacientes que apresentam LLC B típica podem ser subdivididos em três grupos prognósticos maiores. Dois populares sistemas de estadiamento foram desenvolvidos para evidenciar esses grupos prognósticos (Tab. 60.3). Duas outras características podem ser utilizadas para avaliar o prognóstico na LLC B, mas nenhuma tem sido incorporada na classificação de estadiamento: a expressão citoplasmática da proteína ZAP-70, geralmente em linfócitos T, e a expressão de CD38, ambas relacionadas a um pior prognóstico.
Poucas síndromes podem assemelhar-se à LLC (Quadro 60.1). As infecções virais que causam linfocitose podem ser de constatação óbvia devido a febre e outros achados clínicos. No entanto, pode haver febre em casos de LLC por uma infecção bacteriana concomitante. A coqueluche pode causar uma contagem de linfócitos particularmente alta. Outros distúrbios linfoproliferativos, como a macroglobulinemia de Waldeström, a leucemia de células cabeludas ou linfoma, especialmente de células do manto, em fase leucêmica são distinguíveis com base na morfologia, no imunofenótipo dos linfócitos circulantes e na medula óssea. É necessário um hematopatologista experiente para realizar esse diagnóstico diferencial.
A maioria dos casos de LLC inicialmente indolente não requer tratamento específico, e o cuidado-padrão para estágios iniciais da doença tem sido a observação. O tratamento é indicado para pacientes com fadiga progressiva, linfadenopatia sintomática, anemia ou trombocitopenia. Esses pacientes apresentam ou doença sintomática e progressiva estágio Rai II, ou doença estágio III/IV, e o tratamento inicial de escolha é uma combinação de fludarabina (25 mg/m2, dias 2-4, no ciclo 1, e 1-3 nos ciclos subsequentes) e rituximabe (375 a 500 mg/m2 dia 1), com ou sem a adição de ciclofosfamida (250 mg/m² com fludarabina). O clorambucil, 0,6 a 1 mg/kg, via oral, a cada três semanas, por cerca de seis meses, foi o tratamento-padrão antes da fludarabina, sendo conveniente, bem tolerado e permanecendo uma escolha razoável para pacientes idosos que têm dificuldade para deslocar-se ao centro de tratamento. A fludarabina é administrada habitualmente por via IV e é associada à significativa imunossupressão, mas é o agente mais ativo e o único fármaco associado a significativas taxas de remissão completa.
Muitos pacientes que apresentam linfoma podem receber um regime de quimioterapia de combinação utilizado em outros linfomas, como CVP (ciclofosfamida, vincristina e prednisona) ou CHOP (ciclofosfamida, doxorrubicina, vincristina e prednisona), embora regimes contendo fludarabina possam ser preferíveis. Pacientes com anemia hemolítica autoimune ou trombocitopenia imune associadas a essa doença podem necessitar de tratamento com rituximabe, prednisona ou esplenectomia. O uso de fludarabina deve ser evitado em pacientes com anemia hemolítica autoimune, pois pode agravar essa condição, mas a administração concomitante de rituximabe auxilia na redução desse risco.
Os pacientes jovens podem realizar transplante de medula óssea, podendo o alogênico ser curativo, mas associado a altas taxas de mortalidade relacionadas ao tratamento. Minitransplantes que utilizam doses imunossupressoras mais do que doses mieloablativas estão sendo estudados. A realização do transplante de medula óssea autólogo tem sido desencorajada.
As novas terapias estão mudando o prognóstico da LLC. Os pacientes em estágio 0 ou I apresentam uma sobrevida média de 10 a 15 anos e têm relatado que podem ter uma vida normal por muitos anos. Os pacientes em estágio III ou IV evidenciam, com terapias combinadas com fludarabina, uma sobrevida de 2 anos em mais de 90% dos casos, e a perspectiva a longo prazo parece ter mudado substancialmente. Para os pacientes com LCC de alto risco e formas resistentes, há evidência de que o transplante alogênico pode superar os fatores de risco e levar ao controle da doença a longo prazo (Quadro 60.2).
A paciente apresentou significativo prejuízo do estado geral no diagnóstico, com infiltração da medula óssea, lesões dermatológicas, hepatoesplenomegalia e acentuada leucocitose com morfologia típica de “células em flor”. Esses achados clínicos e laboratoriais possibilitaram o diagnóstico de leucemia/linfoma de células T do adulto (LLTA).
A LLTA é um distúrbio linfoproliferativo de linfócitos T maduros associado à infecção pelo vírus linfotrópico de células T humanas tipo I (HTLV-I), uma doença que apresenta polimorfismo clínico e laboratorial. Os pacientes com LLTA apresentam significativa imunodeficiência devido à disfunção dos linfócitos T, a qual favorece a ocorrência de diversas infecções oportunistas.
Com base no diagnóstico, a quimioterapia de indução foi iniciada e obteve resposta hematológica adequada e rápida involução das adenomegalias e da hepatoesplenomegalia. As lesões do escalpo foram tratadas com xampu contendo dexametasona e cetoconazol, com resposta insatisfatória. A paciente desenvolveu infecções virais e fúngicas, mas apresentou recuperação e manteve-se em remissão completa 14 meses após o diagnóstico.
A alta prevalência de infecção pelo HTLV-I, no Brasil, em contraste com a frequência relativamente baixa de LLTA, sugere que esta seja subdiagnosticada. Essa situação pode ser minimizada pelo uso sistemático de imunofenotipagem em todos os casos com um diagnóstico preliminar de linfoma ou leucemia.
1. Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon: IARC; 2008.
Hoffman R, Benz EJ Jr, Shattil Sj, Furie B, Cohen HJ, Silberstein LE, editors. Hematology: basic principles and practice. 5th ed. Philadelphia: Elsevier; 2009.
Linker CA, Damon LE. Blood disorders. In: McPhee SJ, Papadakis MA, Rabow MW, editors. Current: medical diagnosis and treatment. 50th ed. New York; McGraw-Hill; 2011. p. 470-514.
Longo DL.Malignancies of lymphoid cells. In: Kasper D, Fauci A, Longo DL, Braunwald E, Hauser SL, Jameson JL, et al., editors. Harrison´s principles of internal medicine. 17th ed. New York: McGraw-Hill; 2008.p.687-700.
Olivo RA, Martins FF, Soares S, Moraes-Souza H. Adult T-cell leukemia/ lymphoma: report of two cases. Rev Soc Bras Med Trop. 2008;41(3):288-92.
Onciu M. Acute lymphoblastic leukemia. Hematol Oncol Clin North Am.2009;23(4):655-74.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.