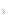 (Carregando Índice)... (Carregando Índice)... |
Autor:
Maria Teresa Correia Caleiro
Médica Assistente do Serviço de Reumatologia do Hospital das Clínicas da Faculdade de Medicina da USP
Última revisão: 22/07/2009
Comentários de assinantes: 0
A doença mista do tecido conectivo (DMTC) foi descrita em 1972 com base em estudos com soro de pacientes que continham altas taxas de anticorpo anti-ribonucleoproteína (anti-RNP) dirigidos a um antígeno extraível do núcleo, a ribonucleoproteína. O conceito original a caracteriza como uma entidade clínica com manifestações de lúpus eritematoso sistêmico (LES), esclerose sistêmica (ES), dermatopolimiosite (DPM) e pela presença do anti-RNP. Os principais achados dessa doença difusa costumam ser edema de mãos, artrite, fenômeno de Raynaud, miosite inflamatória e esclerodactilia.
Segundo os conceitos atuais, a doença engloba manifestações de cinco outras, principalmente LES, esclerodermia, polimiosite, dermatomiosite e artrite reumatóide. Manifestações de síndrome de Sjögren também podem ocorrer, mas caso se dêem isoladamente, mesmo com altos títulos de anti-RNP, a nomenclatura denomina que esses pacientes apresentam síndrome de Sjögren primária. Pacientes com manifestações de mais de uma síndrome reumatológica, mas sem altos títulos de anti-RNP, são rotulados como apresentando síndrome de sobreposição ou overlap.
A etiologia da DMTC é desconhecida e pouco se sabe sobre sua fisiopatologia. Conhecem-se ou especulam-se suas causas devido a semelhanças com ES, LES e DPM, assim como pelas manifestações articulares semelhantes a doença reumatóide (DRe) em grande parte dos pacientes
Com estudos necroscópicos e pesquisas isoladas órgão-específicas, obtiveram-se até agora informações sobre moléculas e células envolvidas na doença, porém nenhuma evidência concreta em relação à fisiopatologia. Os estudos anatomopatológicos dos diferentes tecidos revelam tanto fibrose tecidual como processo inflamatório.
A população mais acometida são mulheres na terceira ou quarta décadas de vida. De início insidioso, as manifestações clínicas mais frequentes na apresentação da doença são fenômeno de Raynaud, poliartrite e fraqueza, presentes em 75% dos casos. Sintomas sistêmicos como febre, perda de peso e adinamia também são comuns. Disfagia baixa é achado também frequente, mas habitualmente não é queixa espontânea dos pacientes, devendo ser interrogada ativamente; pode ou não vir acompanhada de pirose retroesternal e sintomas de refluxo gastroesofágico. Olhos e boca seca (síndrome sicca) podem estar presentes.
Do ponto de vista de antecedentes pessoais e familiares, apenas encontra-se maior prevalência de doenças tiroidianas que na população geral e pode-se verificar a presença de outras doenças auto-imunes na família.
Ao exame do paciente pode-se encontrar linfadenopatia generalizada, frequentemente não referida pelo paciente, hiperemia ocular, que pode estar relacionada a olho seco, e rash cutâneo. O rash pode ser malar ou em outras áreas expostas, como no LES, assim como de localização semelhante à da dermatomiosite, estas mais raras, todas fotossensíveis. Espessamento cutâneo distal às articulações metacarpofalângicas é frequente e encontrado em aproximadamente 75% dos pacientes na apresentação. O espessamento em geral é leve e independente do edema difuso de dedos ou mãos, também muito prevalente. O fenômeno de Raynaud é muito comum e costuma ser bastante intenso, e a capilaroscopia costuma ser similar à dos pacientes com esclerodermia. Já vasculite cutânea e nódulos de subcutâneo são mais raros.
O quadro articular costuma ser simétrico e localiza-se habitualmente em mais de cinco articulações, grandes e pequenas, com franca artrite. Os pacientes desenvolvem artrite erosiva com fator reumatóide (FR) positivo mais frequentemente que no LES. De caráter semelhante ao da DPM, a fraqueza é proximal e de cinturas, com redução de força muscular e reflexos preservados.
O acometimento pulmonar (doença intersticial e fibrose) é muitas vezes silencioso, mas pode se apresentar de forma grave. Quando o paciente tem sintomas, estes serão de dispnéia e tosse seca com ausculta de estertores crepitantes em bases pulmonares. A dismotilidade esofágica pode ser a causa desses achados pulmonares e o exame físico não fará o diagnóstico diferencial. A dismotilidade esofágica, com hipo ou aperistalse, inicia-se em terços distais e acomete posteriormente todo o corpo do órgão. O esfíncter inferior do esôfago é hipotenso e favorece o refluxo.
Ao exame físico, a ausculta cardíaca cuidadosa é fundamental para identificar hipertensão pulmonar, a maior causa de óbito. Nesses casos será encontrada hiperfonese de segunda bulha em foco pulmonar e exame pulmonar normal.
Manifestações renais (glomerulonefrites) e neuropsiquiátricas (psicose e convulsões) são raras, mesmo que haja outras manifestações de LES. Manifestações cardíacas também são raras, sendo a pericardite a mais frequente entre elas.
Exames gerais trazem informações importantes como citopenias no hemograma (todas as séries podem ser afetadas) e elevação das provas de atividade inflamatória não só agudas (VHS, PCR, alfa 2 e beta) como de doença crônica (gama, que chega a mais que 4 mg/dl e está elevada em até 68% dos doentes). Esses exames, no entanto, isoladamente não afastam outras hipóteses que devem ser levantadas, como doenças infecciosas e neoplásicas. Estas devem necessariamente ser afastadas se houver qualquer suspeita clínica.
A elevação de aldolase e/ou CPK, mesmo sem clínica de miopatia, é importante para auxílio diagnóstico e deve ser pesquisada independente de sintomas. Certamente outras causas devem ser afastadas, como o uso de estatinas, tiroidopatia descompensada e toxoplasmose.
A pesquisa de autoanticorpos é fundamental, já que o diagnóstico final depende da presença do anti-RNP em títulos maiores que 1/1.000 por hemaglutinação. Também é necessária a negatividade dos anticorpos anti-DNA de dupla hélice (por imunofluorescência indireta) e do anti-Sm, altamente específicos para LES. O fator antinuclear (FAN) apresenta padrão pontilhado em altos títulos, e ainda mais característico de DMTC que o anti-RNP é a presença de anticorpos contra a proteína 68Kd, que apresenta correlação maior com as manifestações clínicas da doença mista do que o próprio anti-RNP, mesmo porque essa proteína faz parte do complexo RNP. Entretanto, encontrar esse perfil de auto-anticorpos não é suficiente para diagnóstico de DMTC, sendo necessário o conjunto clínico sugestivo e que contemple as indicações para diagnóstico em um dos três critérios de classificação utilizados mundialmente (Tabelas 1 e 2). Os mais utilizados nos últimos cinco anos têm sido os propostos por Kasukawa et al. e o de Alarcon-Segovia, que em um estudo comparativo demonstraram melhor performance diagnóstica.
Tabela 1: Critérios de classificação propostos por Kasukawa et al.
|
1. Sintomas comuns 1.1. fenômeno de Raynaud 1.2. Edema de dedos ou mãos |
|
2. Anticorpos anti-RNP presentes em títulos superiores a 1:1.000 |
|
3. Achados clínicos 3.1. Achados LES-símile: - poliartrite - linfadenopatia - eritema facial - pericardite ou pleurite - leucopenia (< 4.000/mm3) ou trombocitopenia (< 100.000/mm3) 3.2. Achados ESP-símile: - esclerodactilia - fibrose pulmonar, restrição pulmonar ou redução da capacidade de difusão (DCO < 70%) - hipomotilidade ou dilatação do esôfago 3.3. Achados PM-símile: - fraqueza muscular - aumento das enzimas musculares no soro (CPK) - padrão miopático à eletroneuromiografia |
|
Diagnóstico: DMTC será diagnosticada quando todas as três das seguintes condições forem preenchidas: 1. pelo menos um dos sintomas comuns estiver presente; 2. presença de anti-RNP; 3. um ou mais achados em duas ou três das categorias de doença de 3.1, 3.2 e 3.3 |
Tabela 2: Critérios de classificação propostos por Alarcon-Segovia e Villareal
|
1. Sorológico: título de anti-RNP ³ 1:1.600 à hemaglutinação |
|
2. Clínico: Edema de mãos Sinovite Miosite fenômeno de Raynaud Acroesclerose |
|
Diagnóstico: critério sorológico associado a pelo menos três critérios clínicos, incluindo sinovite ou miosite |
A capilaroscopia periungueal pode mostrar quadro compatível com DMTC (padrão SD) em 65% dos pacientes.
Os estudos pulmonares são mandatórios, tanto para diagnóstico da síndrome como para acompanhamento da doença. São necessários (caso disponíveis) tomografia computadorizada de tórax de alta resolução e prova de função pulmonar com volumes e difusão de monóxido de carbono.
O ecocardiograma mostrará a presença ou não de pericardite e poderá mostrar elevação da pressão de artéria pulmonar, que, caso presente, deve ser confirmada por cateterismo e medida direta dos valores pressóricos.
Apesar de sensibilidade de apenas 50%, o estudo radiológico de esôfago com contraste baritado deve ser solicitado e, se normal, deve ser avaliada a necessidade de estudos mais sensíveis como a cintilografia ou a manometria esofágicas em busca de diagnóstico de alterações da motilidade. A endoscopia digestiva alta não dá o diagnóstico de dismotilidade, mas pode mostrar esofagite erosiva que deve ser tratada corretamente para evitar complicações como manifestações extra-esofágicas (p. ex., alterações pulmonares) ou do próprio órgão, como estenoses.
Os diagnósticos diferenciais mais importantes são as outras doenças difusas do tecido conectivo, desde que afastados quadros infecciosos e neoplásicos, como já citado. Dentre essas síndromes, LES, DPM, ES e DRe podem se confundir. Os pacientes, como já comentado, podem apresentar manifestações dessas doenças, mas sem altos títulos de anti-RNP; nesse caso, os pacientes são tidos como portadores de síndrome de sobreposição.
O algoritmo abaixo apresenta um exemplo, tendo o fenômeno de Raynaud como forma de apresentação de um paciente hipotético, apenas para demosntrar como LES, ES, DPM e DMTC podem se relacionar na análise do caso em termos de diagnóstico diferencial, tornando necessária a valorização de todas as variáveis em conjunto. Por serem muitas variáveis, o algoritmo não está completo, pois seria muito longo e outras manifestações igualmente importantes não estão representadas.
Baseia-se no tratamento das outras doenças difusas do tecido conectivo, já que não há trabalhos com grande número de pacientes na literatura ocidental que tornassem possível a orientação terapêutica com guidelines.
Com base em experiência de nosso serviço, citamos que a dupla cloroquina + colchicina é uma boa opção terapêutica em casos leves com artrite + espessamento cutâneo. O uso de drogas citotóxicas é frequentemente necessário, associado ou não à corticoterapia, habitualmente em doses baixas a moderadas. A introdução de gamaglobulina endovenosa está indicada em casos graves e de difícil manejo. Não temos experiência com uso de drogas biológicas.
A hipertensão pulmonar merece atenção especial pela sua gravidade. A melhora da sobrevida tem sido obtida pelo uso de novas drogas como os antagonistas e inibidores de receptores da endotelina, inibidores da fosfodiesterase e análogos da prostaciclina, entre outros, em geral associados à anticoagulação. Em nosso meio, temos obtido boa resposta com a associação de anticoagulação e infusão mensal endovenosa de ciclofosfamida.
A DMTC caracteriza pacientes com altos títulos de anti-RNP e manifestações principalmente de cinco doenças: LES, esclerodermia, polimiosite, dermatomiosite e artrite reumatóide.
A maior parte dos pacientes é constituída por mulheres na 3ª ou 4ª décadas de vida.
Pacientes apresentam fenômeno de Raynaud na maioria dos casos na manifestação da doença, e a capilaroscopia é similar a dos pacientes com esclerodermia.
Raramente ocorrem complicações renais ou neurológicas similares a do LES.
O exame radiológico simples de mãos e punhos dá informações sobre dano ósseo, dirigindo a terapêutica específica, se for o caso.
O diagnóstico de DMTC depende da presença de altos títulos do anti-RNP combinados com manifestações clínicas compatíveis
A capilaroscopia é alterada e compatível com o diagnóstico (padrão SD) em cerca de 65% dos pacientes.
Exames diagnósticos devem ser realizados para detectar complicações pulmonares e esofágicas, mesmo na ausência de sintomas relacionados a esses órgãos.
A doença tem manifestações de diferentes síndromes reumatológicas e apresenta estas como diagnóstico diferencial principalmente.
Altos títulos de anti-RNp diferenciam esses pacientes daqueles com síndrome de sobreposição.
Pacientes com artrite e espessamento cutâneo se beneficiam da combinação de cloroquina e colchicina.
A hipertensão pulmonar beneficia-se com uso de medicações como os inibidores da fosfodiesterase e em nosso meio anticoagulação e ciclofosfamida mensal parece ser particularmente útil.
Algoritmo 1: Avaliação diagnóstica de suspeita de DMTC

1. Amigues JM, Cantagrel A, Abbal M, Mazieres B. Comparative study of four diagnostic criteria sets for MCTD in patients with anti-RNP antibodies. J Rheumatol 1996; 23:2055-62.
2. Aringer M, Steiner G, Smolen JS. Does mixed connective tissue disease exist? Yes. Rheum Dis Clin N Am 2005; 31:411-20.
3. Diógenes Ahm, Bonfá E, Fuller R, Correia Caleiro MT. Capillaroscopy in MCTD: a dynamic process associated with lung involvement and disease severity. Lupus 2007; 16(4):254-8.
4. Kasukawa R. Mixed connective tissue disease. Intern Med 1999; 38(5):386-93.
5. Kim P, Grossman JM. Treatment of mixed connective tissue disease. Rheum Dis Clin N Am 2005; 31:549-65.
6. Klippel JH, Dieppe PA. Rheumatology. 2. ed. Londres: Mosby, 1998.
7. Lundberg IE. The prognosis of mixed connective tissue disease. Rheum Dis Clin N Am 2005; 31:535-47.
8. Pope JE. Other manifestations of mixed connective tissue disease. Rheum Clin N Am 2005; 31:519-33.
9. Sharp GC. MCTD: a concept which stood the test of time. Lupus 2002; 11:333-9.
10. Sharp GC, Hoffman RW. Clinical, immunologic, and immunogenetic evidence that mixed connective tissue disease is a distinct entity: comment on the article by Smolen and Steiner. Arthritis Rheum 1999; 42(1):190-1.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.