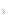 (Carregando Índice)... (Carregando Índice)... |
Autores:
Ricardo Alberto Moreno
Coordenador Geral do Grupo de Estudos de Doenças Afetivas (GRUDA) do Instituto de Psiquiatria do Hospital das Clínicas da Faculdade
de Medicina da Universidade de São Paulo (IPq-HC-FMUSP). Professor Colaborador do Departamento de Psiquiatria da FMUSP.
Doris Hupfeld Moreno.
Mestre e Doutora pelo Departamento de Psiquiatria da FMUSP. Médica Assistente do Grupo de Estudos de Doenças Afetivas (GRUDA)
do IPq-HC-FMUSP.
Ana Claudia Almeida Taveira
Médica Pós-graduanda do Departamento de Psiquiatria da FMUSP. Colaboradora do Grupo de Estudos de Doenças Afetivas (GRUDA)
do IPq-HC-FMUSP.
Última revisão: 01/07/2014
Comentários de assinantes: 0
A depressão é considerada uma doença de bom prognóstico, apesar da propensão à recorrência e à cronicidade. Entretanto, de 25 a 45% dos pacientes tratados com medicação antidepressiva não conseguem atingir remissão.1 Um terço dos pacientes apresentam-se cronicamente sintomáticos2 e 50%, com depressão maior, necessitando de tratamento adicional.3
Os antidepressivos foram descobertos casualmente. No início da década de 1950, investigadores notaram que pacientes com tuberculose apresentavam elevação prolongada do humor quando tratados com iproniazida, um inibidor da monoaminoxidase (IMAO) prescrito como tuberculostático. A iproniazida mostrouse ineficaz no tratamento da tuberculose, mas seu impacto no humor levou a alguns dos primeiros estudos duplo-cegos em psicofarmacologia, demonstrando que os IMAOs eram eficazes na depressão. As observações biológica e farmacológica de que os IMAOs eram antidepressivos e de que a monoaminoxidase degradava a noradrenalina e a serotonina tornaram-se a pedra angular da teoria monoaminérgica da depressão.4
Também foi ao acaso a descoberta posterior de que a imipramina, investigada inicialmente como tratamento para esquizofrenia, elevava o humor, apesar de não ter ação antipsicótica. A descoberta dos antidepressivos e sua utilização na prática clínica trouxeram um avanço importante no tratamento e no entendimento de possíveis mecanismos subjacentes aos transtornos depressivos,1,4 o que tornou a depressão um problema médico passível de tratamento, semelhante a outras doenças, como o diabete e a hipertensão arterial.
Até a década de 1980, havia duas classes de antidepressivos: os tricíclicos (ADTs) e os inibidores de monoaminoxidase (IMAOs). Embora muito eficazes, essas drogas apresentavam efeitos colaterais indesejáveis, causados pela inespecificidade de sua ação farmacológica, e eram potencialmente letais em casos de superdosagem. Nas últimas duas décadas, surgiram novas classes de antidepressivos a partir da pesquisa de moléculas desprovidas dos efeitos colaterais dos heterocíclicos. Os novos antidepressivos diferem dos clássicos ADTs e IMAOs, irreversíveis pela seletividade farmacológica, modificando e atenuando os efeitos colaterais.4 Além disso, apresentam um perfil reduzido de interações medicamentosas, constituindo importantes opções no tratamento de episódios depressivos.
O objetivo do tratamento antidepressivo é a remissão dos sintomas, o que diminui o risco de recorrência, melhora a qualidade de vida, melhora a capacidade funcional e indica um bom prognóstico.5,6
O tratamento da depressão é dividido em três fases (Fig. 5.1)7: aguda, de continuação e de manutenção. A fase aguda visa à remissão dos sintomas (ausência de sintomas ou sinais para preencher o diagnóstico de transtorno) e ao início da recuperação do funcionamento psicossocial. Sua duração varia de 6 a 12 semanas, e o paciente assintomático por seis meses é considerado como recuperado do episódio atual. A fase de continuação visa à prevenção de recaídas (retorno dos sintomas do episódio índice). Sua duração varia de 4 a 9 meses. O tratamento de manutenção está indicado para aos pacientes com grande risco de recorrência ao longo da vida: depressões crônicas (duração acima de dois anos), episódios graves (com tentativas de suicídio ou com sintomas psicóticos), depressões resistentes a tratamento (mais de dois episódios em dois anos), depressões recorrentes (três ou mais episódios ao longo da vida) e depressão na velhice (acima dos 65 anos).8 Deve-se estender o tratamento por um ou mais anos e, em alguns casos, este deve ser vitalício.Recomenda-se manter, durante todo o tratamento, a dose necessária de medicamento para atingir a remissão dos sintomas.8,9
A resposta ao tratamento é medida pela melhora clínica do paciente (sintomatológica e funcional) e pode ser parcial ou total. O objetivo do tratamento é a remissão dos sintomas e a recuperação dos níveis normais de funcionamento e de bem-estar do paciente, pois a manutenção de sintomas residuais agrava o prognóstico. Diante de uma resposta parcial, a primeira conduta é o aumento da dose; se não houver remissão, troca-se o medicamento por outra classe de antidepressivos. Em caso de não haver resposta novamente, deve-se considerar a possibilidade de depressão resistente a tratamento (DRT).9
Hoje dispomos de mais de 30 compostos antidepressivos distintos (Tab. 5.1). A escolha do antidepressivo deve estar embasada nas evidências de eficácia do medicamento (Quadro 5.1),6,10-17 nas características clínicas do episódio depressivo, no perfil de efeitos adversos e na história pessoal ou familiar de resposta anterior a determinada medicação. Além disso, a interação medicamentosa e seu uso em populações específicas (criança, idoso, gravidez, ou associado a outras comorbidades clínicas) também devem ser considerados nessa escolha. Para uma boa prática clínica, recomenda-se usar o antidepressivo em dose terapêutica (dose máxima indicada pela posologia) e reavaliar sua continuidade após 3 semanas, caso não haja nenhum tipo de resposta. O tempo adequado de tratamento varia de 6 a 12 semanas, e, em caso de retirada da medicação, esta deve ser gradual, para evitar o aparecimento de sintomas de descontinuação abrupta.

Figura 5.1
As fases do tratamento da depressão. (Adaptada de Kupfer7)
Tabela 5.1
Quadro 5.1
Em casos de DRT, recomenda-se a reavaliação psiquiátrica e médica, com objetivo de identificar comorbidades clínicas e/ou psiquiátricas (Quadro 5.2).8,9 A avaliação psiquiátrica deve incluir a cronologia dos sintomas, sua duração e a resposta aos tratamentos anteriores. O erro diagnóstico entre depressão unipolar e bipolar contribui para resposta pobre ao tratamento. Exames clínicos como hemograma, avaliação hepática e renal, ECG, teste de gravidez (quando for o caso) e exames de eletrólitos e de hormônios tireoidianos são necessários. Muitas vezes, o paciente abandona o tratamento na fase aguda, e é importante lembrá-lo de riscos e de benefícios da medicação, bem como do curso da doença depressiva, visando a aumentar a adesão ao tratamento.
Algumas estratégias podem ser utilizadas para aumentar a eficácia antidepressiva em pacientes com resposta parcial. Uma delas é aumentar a dose do antidepressivo em uso (algumas vezes em níveis superiores aos sugeridos na posologia, conforme tolerabilidade e eficácia). Para atingir nível plasmático adequado em metabolizadores rápidos, é imperativa a prescrição de doses adequadas. Doses baixas podem produzir efeito serotoninérgico específico, enquanto doses altas promovem ação dual, como se observa com a venlafaxina. O aumento gradual da dose inclui titulação de 50 a 100% da dose a cada 3 a 7 dias, dependendo da resposta e dos efeitos adversos. Recomenda-se aguardar, no mínimo, quatro semanas para avaliar a eficácia da substância. Em caso de não resposta ou de resposta insatisfatória, recomenda-se trocar o medicamento por outro antidepressivo de outra classe farmacológica. Outras estratégias incluem a adição de um antidepressivo de outra classe ou a potencialização do efeito antidepressivo na fase aguda do tratamento de pacientes que não responderam satisfatoriamente à monoterapia. As substâncias mais estudadas na potencialização são o lítio e a triiodotironina (T3). Os antipsicóticos atípicos também têm se mostrado eficazes na potencialização, com diminuição da ansiedade e da agitação.3,9,21,22
Quadro 5.2
Os antidepressivos podem ser classificados de acordo com sua estrutura química ou com suas propriedades farmacológicas. A estrutura cíclica (anéis benzênicos) caracteriza os antidepressivos heterocíclicos (tricíclicos e tetracíclicos). Os tricíclicos (ADTs) se dividem em dois grandes grupos: as aminas terciárias (imipramina, amitriptilina, trimipramina e doxepina) e as aminas secundárias (desmetilimipramina, nortriptilina e protriptilina). Maprotilina e amoxapina são antidepressivos tetracíclicos. As características farmacológicas da maprotilina se assemelham às dos ADTs, e essa droga será abordada dentro dessa classe de antidepressivos. Atualmente, os antidepressivos são classificados, preferencialmente, em função da ação farmacológica, classificação mais útil na prática clínica, porque os antidepressivos de nova geração não compartilham estruturas comuns. Podemos dividi-los de acordo com o mecanismo de ação proposto, aumentando a eficiência sináptica da transmissão monoaminérgica (particularmente de neurônios noradrenégicos e/ou serotoninérgicos) (Fig. 5.2).13,23,24 Medicamentos antidepressivos produzem aumento na concentração de neurotransmissores na fenda sináptica por meio da inibição do metabolismo, do bloqueio da recaptação neuronal ou da atuação em auto-receptores pré-sinápticos (Tab. 5.1).
Os IMAOs são bem absorvidos pelo trato gastrintestinal, sofrem biotransformação hepática rápida por oxidação e, possivelmente, têm metabólitos ativos. O início de ação se dá entre 7 e 10 dias em alguns pacientes com doses apropriadas, mas pode levar de 4 a 8 semanas para atingir o efeito terapêutico pleno. O pico de concentração plasmática é de 3 a 5 horas para isocarboxazida, 2 a 4 para fenelzina e 1 a 3,5 para tranilcipromina. Em média, são necessários 10 dias para que a atividade da MAO se recupere, já que em 5 a 10 dias os IMAOs irreversíveis inibem as MAOs A e B de forma permanente. Elas voltam a ser produzidas em uma a duas semanas, mas nessa fase o paciente continua vulnerável ao desencadeamento de crises hipertensivas pelo aumento da concentração de aminas provenientes da dieta ou de medicamentos aminérgicos. A eficácia da fenelzina se correlaciona com a inibição de 80% da MAO plaquetária, ao passo que o melhor preditor de resposta terapêutica da tranilcipromina parece ser a área sobre a curva cinética. A eliminação é renal, inclusive dos metabólitos. A moclobemida inibe apenas a MAO-A, por tempo menos prolongado (aproximadamente 24 horas apenas) e de forma reversível. Consequentemente, não é necessário aguardar duas semanas até que a MAO volte a ser produzida e outros antidepressivos possam ser prescritos.24,25,27
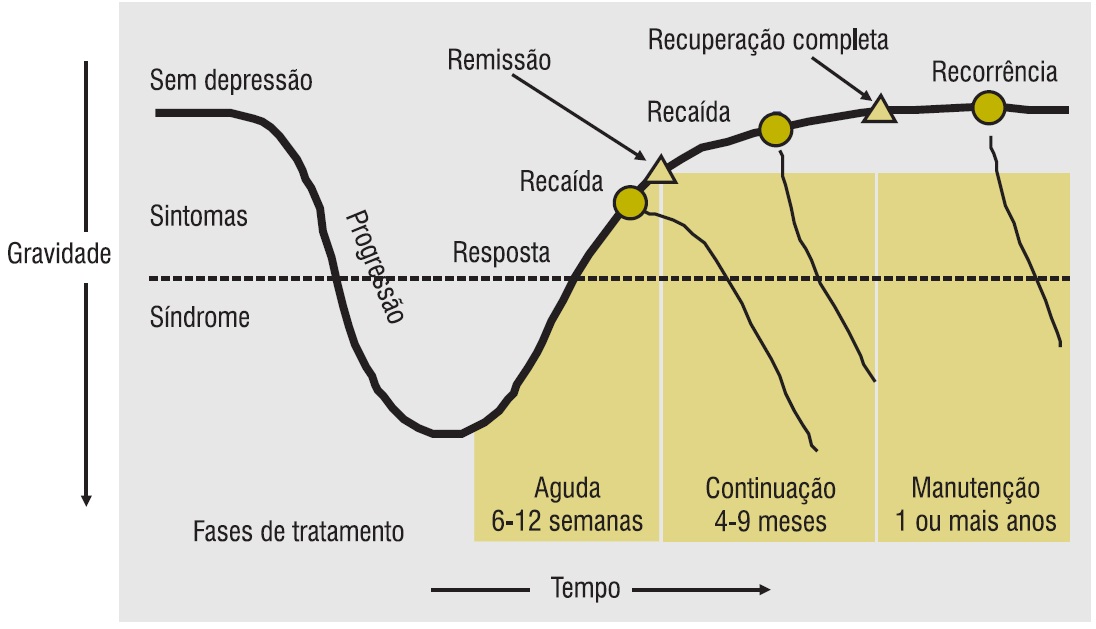
Figura 5.2
Seletividade de inibição da recaptação in vitro de antidepressivos. (Adaptada de Bezchlibnyk-Butler; Jeffries25; Feighner26; Kaplan; Sadock24.)
O mecanismo de ação dos IMAOs foi pouco estudado e ainda não está totalmente esclarecido.28 Sabe-se que a atividade da enzima monoaminoxidase (MAO) está inibida. Os subtipos da MAO, A e B, estão envolvidos no metabolismo de serotonina, noradrenalina e dopamina. Isocarboxazida, fenelzina e tranilcipromina são IMAOs não seletivos que se ligam de forma irreversível às MAOs. A redução na atividade da MAO resulta em aumento na concentração desses neurotransmissores nos locais de armazenamento no sistema nervoso central (SNC) e no sistema nervoso simpático. O incremento na disponibilidade de um ou mais neurotransmissor tem sido relacionado à ação antidepressiva dos IMAOs. A inibição não seletiva dos IMAOs fenelzina, isocarboxazida e tranilcipromina resulta em subsensibilização de receptores a-2 ou ß-adrenérgicos e de serotonina. Possivelmente, as mudanças nas características dos receptores produzidas pela administração crônica de IMAOs correlacionam-se melhor com a atividade antidepressiva do que o aumento na atividade do neurônio secundária ao aumento na concentração de neurotransmissores, o que pode também explicar a demora para o início da ação terapêutica. Mais recentemente, foram desenvolvidos IMAOs seletivos da MAO-A e da MAO-B, além de compostos reversíveis, que contornam o problema das crises hipertensivas (Tab. 5.1). A moclobemida é um antidepressivo inibidor seletivo da MAO-A e reversível, que desamina 5-HT e NA, ao passo que inibidores seletivos da MAO-B, como a selegilina, não possuem ação antidepressiva significativa.
O efeito adverso mais comumente descrito é a hipotensão postural. Esse efeito pode ser diminuído com início gradual da medicação (Tab. 5.2).
Tabela 5.2
Pode ocorrer na associação com outros antidepressivos (amitriptilina, clomipramina, doxepina, imipramina, ISRSs ou trazodona). Essa síndrome também pode ocorrer na substituição entre substâncias, quando não se observa período de wash-out adequado para a total eliminação da substância. Foram descritas alterações da cognição e do comportamento (confusão, hipomania, agitação), do sistema nervoso autônomo (diarreia, febre, diaforese, efeitos na pressão arterial, náuseas e vômitos) e do sistema neuromuscular (mioclonias, hiperreflexia, incoordenação e tremores). A melhora é rápida com a retirada das substâncias.
Pelo fato de os IMAOs inibirem a MAO de forma permanente, é necessário adotar uma dieta pobre em tiramina, aminoácido precursor de catecolaminas, de modo a evitar uma crise hipertensiva potencialmente fatal.
Na Tabela 5.329 encontra-se a lista de alimentos e medicamentos proibidos e permitidos, utilizada no Grupo de Estudos de Doenças Afetivas, do Instituto de Psiquiatria do Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo (GRUDA).
Tabela 5.3
Os sintomas da crise hipertensiva decorrentes da interação de IMAOs com substâncias ricas em tiramina ou em aminas biogênicas são cefaleia intensa, palpitações, dor torácica intensa, dilatação das pupilas, taquicardia ou bradicardia e aumento da fotossensibilidade. Pode haver aumento da sudorese, febre ou sensação de frio, pele viscosa, náusea ou vômitos e rigidez da nuca. Existem relatos de hemorragia intracraniana (algumas vezes fatal) em consequência das crises hipertensivas. Palpitação ou cefaleia frequente constituem sintomas prodrômicos da reação hipertensiva.9,30
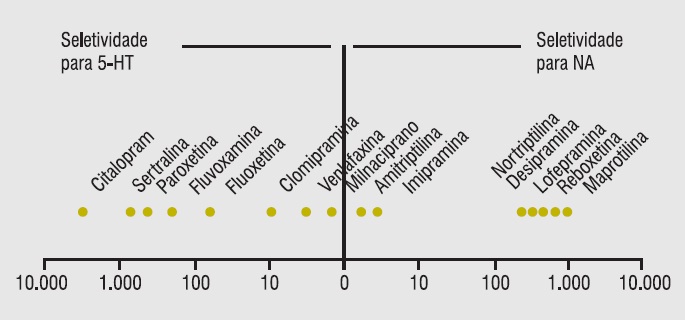
Na Tabela 5.4 estão descritas as principais interações entre IMAOs e outros medicamentos.
Tabela 5.4
Os ADTs são bem absorvidos pelo trato gastrintestinal e metabolizados em grande parte (55 a 80%) pelo efeito de primeira passagem no fígado. O pico plasmático é atingido mais rapidamente (1 a 3 horas) por aminas terciárias (como a amitriptilina) do que por aminas secundárias (desipramina e nortriptilina), que levam de 4 a 8 horas para atingi-lo. São altamente lipofílicos, concentrando-se principalmente no miocárdio e nos tecidos cerebrais, e ligam-se a proteínas plasmáticas. Muitos ADTs apresentam farmacocinética linear, isto é, mudanças na dose levam a alteração proporcional no nível plasmático. A vida média de eliminação varia (p. ex., imipramina de 4 a 34 horas, amitriptilina de 10 a 46 horas, clomipramina de 17 a 37 horas e nortriptilina de 13 a 88 horas), e o estado de equilíbrio é atingido em cerca de 5 dias. A farmacocinética pode variar entre os sexos, e a concentração pode diminuir antes da menstruação.18,25
O mecanismo de ação comum aos antidepressivos tricíclicos em nível pré-sináptico é o bloqueio da recaptação de monoaminas, principalmente norepinefrina (NE) e serotonina (5-HT) e, em menor proporção, dopamina (DA). Aminas terciárias inibem preferencialmente a recaptação de 5-HT, e aminas secundárias, a de NE (Tab. 5.1). Atualmente, considera-se não haver diferenças significativas quanto à seletividade do bloqueio da recaptação pré-sináptico. A atividade pós-sináptica varia de acordo com o sistema neurotransmissor envolvido e, geralmente, é responsável pelos efeitos colaterais. Os ADTs bloqueiam receptores muscarínicos (colinérgicos), histaminérgicos de tipo 1, a-2 e ß-adrenérgicos, serotoninérgicos diversos e, mais raramente, dopaminérgicos. Essas ações não se correlacionam necessariamente com efeito antidepressivo, mas com efeitos colaterais (Tab. 5.5).9,18,19 O bloqueio do receptor 5-HT1 contribuiria para o efeito terapêutico. Contudo, essa ação aguda dos antidepressivos no sistema de transmissão monoaminérgica, por si só, não explicava a demora para o início da ação antidepressiva, observável clinicamente após duas semanas de uso. Estudos recentes das vias receptoras pós-sinápticas e de mensageiros secundários, assim como da expressão genética, podem desempenhar papel importante na elucidação das mudanças que ocorrem a longo prazo no funcionamento cerebral resultante da utilização crônica de antidepressivos. Embora o mecanismo de ação exato não tenha sido totalmente elucidado, sabe-se que os ADTs promovem, agudamente, aumento na eficiência da transmissão monoaminérgica (e, possivelmente, GABAérgica), envolvendo os sistemas noradrenérgico e serotoninérgico, por meio do aumento da concentração sináptica de norepinefrina e de serotonina por bloqueio da recaptação. Cronicamente, os ADTs dessensibilizam receptores ß-1 adrenérgicos, serotoninérgicos 5-HT2 e, provavelmente, 5-HT1A no sistema nervoso central. Sistemas mensageiros secundários estão envolvidos nessas mudanças.31 AMP cíclico, cálcio, diacilglicerol e fosfolípides estimulam a fosforilação de quinases proteicas, possivelmente envolvidas na síntese de catecolaminas, e podem aumentar a ligação de proteína G a receptores subsequentemente dessensibilizados, exercendo ação reguladora no receptor. Os hormônios (como estradiol e progesterona) são substâncias também envolvidas na alteração da sensibilidade ou no número de receptores pelos ADTs, interferindo na capacidade de ligação da imipramina ao hipotálamo. A ação antienurética do hidrocloridro de imipramina não está estabelecida. Acredita-se que esteja associada com o efeito anticolinérgico da imipramina. O efeito antiobsessivo da clomipramina talvez se correlacione com a inibição da recaptação de serotonina e com a consequente subsensiblização compensatória dos subtipos de receptores serotoninérgicos. No transtorno do pânico, os estudos sugerem prejuízo no funcionamento do sistema nervoso autônomo, o que causa liberação excessiva de norepinefrina do locus ceruleus. Pensa-se que os ADTs diminuam a taxa de disparo do locus ceruleus por regulação na função de receptores a-2 e ß-adrenérgicos e no turnover de noradrenalina. A ação antinevrálgica dos ADTs não está necessariamente relacionada à melhora da depressão. A analgesia pode ser mediada por mudanças na concentração central de monoaminas, particularmente serotonina, e também pelo efeito direto ou indireto dos ADTs nos sistemas opioides endógenos. Na úlcera péptica, os ADTs são eficazes na melhora da dor. Eles ajudam na cicatrização completa pela sua capacidade de bloquear receptores H2 nas células parietais e pelo seu efeito sedativo e anticolinérgico. Na bulimia nervosa, parece haver efeito independente da melhora da depressão. O mecanismo de ação envolvido na incontinência urinária pode incluir atividade anticolinérgica, resultando no aumento da capacidade vesical, na estimulação diretaa-adrenérgica e na atividade agonista a-adrenérgica, no aumento do tônus esfincteriano e também no bloqueio central da recaptação. Outras ações dos ADTs incluem efeito anticolinérgico periférico e central, devido à potente e elevada afinidade de ligação por receptores muscarínicos, efeito sedativo, pela forte afinidade de ligação por receptores histamina H1, e efeito de hipotensão ortostática, devido ao bloqueio a-adrenérgico. Além disso, os ADTs são agentes antiarrítmicos da classe 1A que, como a quinidina, em doses terapêuticas, diminuem moderadamente a condução intraventricular e, em doses elevadas, podem causar bloqueio grave de condução e arritmias ventriculares.31
Os efeitos colaterais dos ADTs estão associados com sua afinidade pelos receptores muscarínicos, histaminérgicos, serotoninérgicos, noradrenérgicos e a-1 adrenérgicos (Tab. 5.5).
Associados ao bloqueio muscarínico, são os efeitos colaterias mais frequentes dos ADTs, e sua intensidade declina com o passar do tempo ou com a redução do antidepressivo. Seus efeitos são boca seca (recomenda-se estimular higiene bucal frequente), visão turva (por dificuldade de acomodação visual), obstipação (em idosos há risco de íleo paralítico) e retenção urinária.
Ocorrem aumento da frequência cardíaca, achatamento da onda T, raramente prolongamento do intervalo PR e aumento do complexo QRS. Geralmente são dose-dependentes e observados em concentrações plasmáticas acima dos níveis terapêuticos. Ocorre hipotensão postural (idosos devem ser orientados e monitorados pelos riscos de quedas, e, nesses casos, a nortriptilina estaria mais indicada). As propriedades antiarrítmicas quinidina-símile dos ADTs favorecem seu uso em pacientes com extrassístoles ventriculares.
Ocorrem tremores de mãos, sedação (principalmente com o uso de amitriptilina e maprotilina), latência para evocar lembranças, dificuldade para encontrar palavras, gagueira, mioclonias, parestesias, agitação e hiperestimulação paradoxal. Estados confusionais podem ocorrer em idosos, mas raramente há ocorrência de convulsões (em caso de doses elevadas ou aumento rápido da medicação, principalmente com maprotilina e clomipramina), movimentos coreoatetoides e acatisia. Os pacientes devem ser orientados para não operarem máquinas perigosas, nem dirigirem veículos (caso estejam sonolentos), e para evitarem consumo de álcool.
Ocorre aumento da secreção de prolactina, mas galactorreia e amenorreia secundária são raras. Outro efeito raro é a hiponatremia da síndrome de secreção inadequada do hormônio antidiurético, que foi descrita com o uso de amitriptilina e clomipramina.
Ocorrem exantemas, urticária, eritema multiforme, dermatite esfoliativa e fotossensibilidade. Esses efeitos ocorrem em 2 a 4% dos pacientes nas duas primeiras semanas de tratamento.
Raramente ocorrem alterações de função hepática.
Não menos importantes, referem-se aos que podem ser confundidos com a própria sintomatologia depressiva. Estão incluídos ganho de peso, associado ou não à preferência por carboidratos, principalmente com amitriptilina e imipramina, disfunções sexuais (redução da libido, retardo ou inibição ejaculatória e inibição do orgasmo) e alterações do sono (pesadelos, alucinações hipnagógicas e hipnopômpicas). O aumento de ansiedade e a síndrome tricíclica precoce podem ocorrer nos primeiros dias de tratamento, principalmente em pacientes com ataques de pânico, e melhoram com associação a benzodiazepínicos. Dificuldades de memória são mais comuns em idosos e no curso do tratamento profilático.
Tabela 5.5
Em um pequeno grupo de pacientes, a interrupção abrupta de ADTs, principalmente após tratamento prolongado, é acompanhada de uma síndrome de descontinuação que ocorre nas primeiras 48 horas após a suspensão do antidepressivo. Os sintomas podem estar relacionados a um efeito rebote de hiperatividade colinérgica. Clinicamente, a síndrome se caracteriza por sintomas de mal-estar geral, alterações gastrintestinais (náuseas, vômitos, diarreia), ansiedade, irritabilidade, insônia, sonhos vívidos, movimentos parkinsonianos ou acatisia. Podem ocorrer ataques de pânico, arritmias cardíacas, delirium e, menos frequentemente, agitação. Recomenda-se a diminuição gradativa da medicação ao longo de algumas semanas. O esquema seguido no GRUDA consiste na retirada imediata de 50% da dose e de 25% do restante a cada dois dias.20
Caracterizada por confusão, convulsões, alterações de concentração, sonolência grave, alargamento de pupilas, alteração da frequência cardíaca, febre, alucinações, inquietação ou agitação, respiração curta ou difícil, cansaço, fraqueza intensa e vômitos. O tratamento da intoxicação consiste em diminuição da absorção do medicamento (esvaziamento gástrico com lavagem), aumento da sua eliminação (administração de pasta de carvão ativado seguida de estimulação catártica) e tratamento específico das intercorrências cardiopulmonares.20
Durante a gestação, é possível utilizar ADTs; preferencialmente, evitando seu uso no primeiro trimestre. Amitriptilina, clomipramina, desipramina e nortriptilina são antidepressivos que, em estudos de reprodução em animais, mostraram algum efeito adverso no feto; em humanos, não há estudos adequados e bem controlados. Contudo, não há relatos de associação significativa entre ADTs e más formações congênitas descritos até o momento, mesmo quando usados no primeiro trimestre da gestação10. Os ADTs devem ser suspensos duas semanas antes do parto, a fim de evitar problemas cardíacos, irritabilidade, desconforto respiratório, espasmos musculares, convulsões ou retenção urinária em neonatos.
Mulheres lactantes podem tomar ADTs, preferencialmente imipramina e amitriptilina, mas também nortriptilina e clomipramina. A maprotilina deve ser evitada por causa de sua meia-vida longa.
Os ADTs estão contra-indicados nos casos de glaucoma de ângulo fechado. Efeitos na condução cardíaca normalmente não apresentam significado clínico, mas os ADTs são contraindicados em casos de bloqueios de ramo esquerdo, de bloqueio AV total, de alterações na condução intracardíaca e de infarto agudo do miocárdio. O eletrocardiograma constitui um método sensível e deve ser solicitado quando houver suspeita de alterações cardíacas e em pacientes acima de 50 anos.
Há registro de interações medicamentosas de significativa importância entre antidepressivos tricíclicos e outros medicamentos comumente utilizados em idosos:20,23,32
Ø Analgésicos: os ADTs possuem efeito antiálgico, permitindo que doses menores de analgésicos sejam empregadas.
Ø Anestésicos: a administração de halotano e de pancurônio requer cautela pelo efeito anticolinérgico dos ADTs. Recomenda-se o uso de relaxantes musculares sem efeitos vagolíticos e simpatomiméticos.
Ø Agentes anticolinérgicos: a administração conjunta de ADTs e antiparkinsonianos pode levar à potencialização de efeitos atropínicos. Sintomas de síndrome anticolinérgica podem ocorrer, tais como ansiedade, agitação, desorientação, disartria, comprometimento de memória, alucinações, mioclonias, convulsões, taquicardia, arritmias, midríase, elevação da temperatura
corporal, obstipação intestinal e retenção urinária.
Ø Anticoagulantes: relatos de casos isolados sugerem cuidados na interação entre anticoagulantes e antidepressivos tricíclicos, especialmente no que se refere à análise do tempo de protrombina em pacientes que recebem tratamentos combinados.
Ø Anticonvulsivantes: a carbamazepina pode aumentar o metabolismo de imipramina, doxepina e amitriptilina, reduzindo em 42 a 50% os níveis plasmáticos. ADTs reduzem o limiar convulsígeno e podem comprometer o efeito de barbitúricos. Os níveis plasmáticos da fenitoína podem ser elevados por imipramina, mas não por nortriptilina ou amitriptilina.
Ø Anti-hipertensivos: a guanetidina não deve ser utilizada em pacientes que fizerem uso de antidepressivos bloqueadores da recaptação de norepinefrina, e a clonidina também deve ser evitada; metildopa e diuréticos tiazídicos podem ser empregados, evitando hipotensão e hipocalemia; verapamil e diltiazem podem inibir a metabolização de imipramina por interação no sistema citocromo P450, podendo ser necessário reduzir a dose do antidepressivo.
Ø Bloqueadores histamínicos H2: a cimetidina pode inibir a metabolização hepática de ADTs, elevando os níveis séricos e o risco de toxicidade. Ela pode aumentar a biodisponibilidade de imipramina, porém não a de nortriptilina. Suspender a cimetidina do paciente em uso crônico de ADTs pode reduzir os níveis séricos terapêuticos. Sugere-se monitoração plasmática ao introduzir e ao retirar cimetidina.
Ø Levodopa: a associação pode ter efeito sinérgico nos sistemas colinérgicos e catecolaminérgicos, aumentando efeitos colaterais.
Ø Quinidina: a associação com desipramina pode aumentar os níveis séricos e o risco de toxicidade.
Ø Reserpina: a reserpina depleta agudamente monoaminas intraneuronais. A associação pode levar a efeitos colaterais como diarreia, vasodilatação cutânea ou mesmo sintomas maniformes. Recomenda-se cuidado nesta combinação.
Ø Aminas simpatomiméticas: a administração de noradrenalina ou outras aminas simpatomiméticas em pacientes recebendo ADTs pode levar a efeito sinérgico, aumentando o tônus simpático.
Os ISRSs (citalopram, fluoxetina, fluvoxamina, paroxetina, sertralina e escitalopram) são o resultado de pesquisa racional para encontrar medicamentos tão eficazes quanto os ADTs, mas com poucos problemas de tolerabilidade e de segurança. Os ISRSs inibem de forma potente e seletiva a recaptação de serotonina, resultando em potencialização da neurotransmissão serotoninérgica. Embora compartilhem o principal mecanismo de ação, os ISRSs são estruturalmente distintos, com marcadas diferenças nos perfis farmacodinâmico e farmacocinético. A potência da inibição da recaptação da serotonina é variada (Fig. 5.2), assim como a seletividade por noradrenalina e dopamina. Sertralina e paroxetina são os mais potentes inibidores da recaptação. A potência relativa da sertralina em inibir a recaptação de dopamina a diferencia farmacologicamente dos outros ISRSs. A afinidade por neurorreceptores, tais como sigma, muscarínicos e 5-HT2c, também difere muito. Mais ainda, a inibição da sintetase óxido-nítrica pela paroxetina, e possivelmente por outros ISRSs, pode ter efeitos farmacodinâmicos significativos. Citalopram e fluoxetina são misturas racêmicas de diferentes formas quirais, que possuem perfil farmacodinâmico e farmacocinético variados. Fluoxetina possui metabólito de ação prolongada e farmacologicamente ativo. Os ISRSs também apresentam perfil farmacocinético variado, que inclui meia-vida, farmacocinética linear versus não linear, efeito da idade no clearance e no seu potencial de inibir isoenzimas metabolizadoras de medicamentos do citocromo P450 (CYP). Essas diferenças farmacológicas e farmacocinéticas sustentam as diferenças clínicas cada vez mais importantes entre os ISRSs.5,28
Embora todos os ISRSs apresentem o mesmo mecanismo de ação, as diferenças entre as estruturas moleculares fazem com que os diferentes compostos apresentem perfis farmacocinéticos diversos. Todos os ISRSs apresentam alta ligação proteica (fluvoxamina e citalopram em menor grau). A fluoxetina é a única que apresenta metabólito com atividade clínica significativa (inibição da recaptação de serotonina e de isoenzimas do citocromo P450), a norfluoxetina. A meia-vida prolongada da fluoxetina e da norfluoxetina e o tempo necessário para se atingir o estado de equilíbrio apresentam significado clínico, como a maior latência para o início da ação antidepressiva. As concentrações plasmáticas de sertralina e de citalopram são proporcionais às doses administradas (farmacocinética linear), o que não ocorre com fluoxetina, paroxetina e fluvoxamina, cuja farmacocinética não é linear. Esses ISRSs diminuem seu metabolismo por ação inibitória dosedependente das isoenzimas do citocromo P450, o que significa que aumentos na dose administrada de fluoxetina, paroxetina e fluvoxamina levam a aumentos desproporcionais nos níveis plasmáticos e nas meias-vidas e, possivelmente, levam a efeitos colaterais.20,22,28,30
Os ISRSs são rapidamente absorvidos, sofrem menos efeito do metabolismo de primeira passagem e ligam-se fortemente a proteínas plasmáticas. Todos (em menor proporção, a fluvoxamina) deslocam outras drogas da ligação proteica, aumentando seu nível plasmático. Metabolizados primariamente pelo fígado, todos os ISRSs afetam as enzimas metabolizadoras do citocromo P450 (em menor proporção, a sertralina) e podem comprometer o metabolismo de outras drogas metabolizadas por esse sistema. Tem-se demonstrado que fluoxetina e paroxetina diminuem seu metabolismo com o tempo. O pico plasmático da sertralina aumenta 30% quando o medicamento é ingerido com alimentos, pela diminuição do metabolismo de primeira passagem.
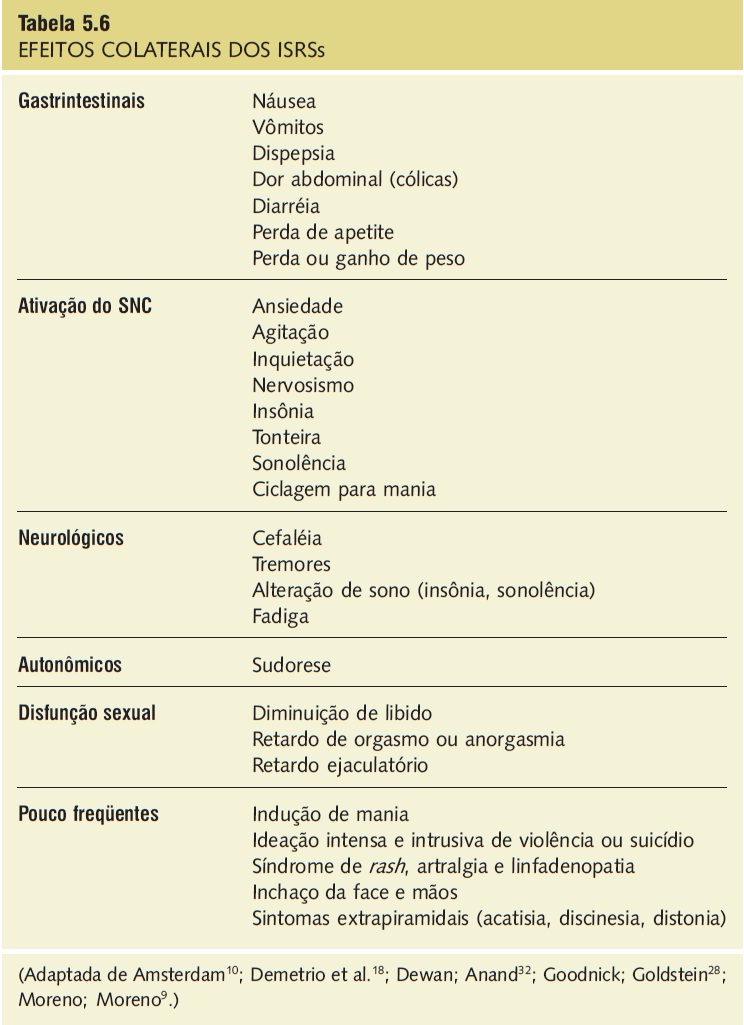
Em função de sua ação seletiva, os ISRSs apresentam perfil mais tolerável de efeitos colaterais, que são diferentes entre os diversos medicamentos. De forma geral, os efeitos colaterais mais frequentemente relatados são de trato gastrintestinal, de ativação do sistema nervoso central (agitação, ansiedade, insônia, ciclagem para mania, nervosismo), de alterações do sono, de fadiga, de ordem neurológica (tremores, efeitos extrapiramidais), de perda ou ganho de peso, de disfunções sexuais e de reações dermatológicas (Tab. 5.6).9,10,18,28,32
Ø Efeitos gastrintestinais: os efeitos anticolinérgicos da paroxetina podem levar a uma maior incidência de obstipação intestinal em vez de diarreia. Por outro lado, alguns estudos sugerem maior incidência de diarréia com a sertralina em relação a fluoxetina e citalopram.
Ø Reações dermatológicas: são mais frequentes com a fluoxetina. Elas aparecem na forma de urticária, que pode estar acompanhada de febre, artralgia e eosinofilia.
Ø Efeitos psiquiátricos: a fluoxetina parece estar mais relacionada ao aparecimento de efeitos colaterais como agitação, insônia, ansiedade, ciclagem para mania e nervosismo. Entretanto, os outros ISRSs podem apresentar os mesmos efeitos com o emprego de doses mais elevadas.
Ø Alterações de peso: a sertralina está associada a uma discreta perda de peso no início do tratamento. A fluoxetina parece ser mais potente na inibição do apetite, com maior perda de peso no início do tratamento. A paroxetina, ao contrário, foi associada a ganho de peso, o que também foi relatado com o citalopram.
Ø Disfunção sexual: o uso de ISRSs foi associado, principalmente, a retardo ejaculatório em homens e a anorgasmia em mulheres. A paroxetina está associada à maior incidência desses efeitos colaterais, o que poderia ser explicado por sua potência na inibição da recaptação de serotonina e por sua mínima atividade dopaminérgica.
Os sintomas que aparecem na retirada dos ISRSs são clinicamente benignos e podem aparecer no período entre 1 e 10 dias após a retirada da medicação (embora no caso da fluoxetina possam aparecer várias semanas depois, em função de seu perfil farmacocinético) e persistir por até três semanas. Os sintomas mais frequentes são tonturas, vertigens, ataxia, sintomas gastrintestinais (náuseas e vômitos), sintomas gripais, distúrbios sensoriais (parestesias), alterações de sono (insônia, sonhos vívidos) e sintomas psíquicos (irritabilidade, agitação, ansiedade). Assim como acontece com outras substâncias psicoativas, esses sintomas podem ser resultantes de alterações adaptativas que mais frequentemente envolvem o ajustamento de receptores para compensar a atividade farmacológica da droga (efeito rebote). O aparecimento dos sintomas correlaciona-se com a queda nos níveis plasmáticos do ISRS, o que explica sua maior incidência na retirada de paroxetina e de fluvoxamina do que durante a retirada de fluoxetina. Entretanto, a maior ocorrência desses sintomas com a paroxetina pode ser explicada não apenas por seu perfil farmacocinético, mas também por seus efeitos anticolinérgicos.9,33,34
Tabela 5.6
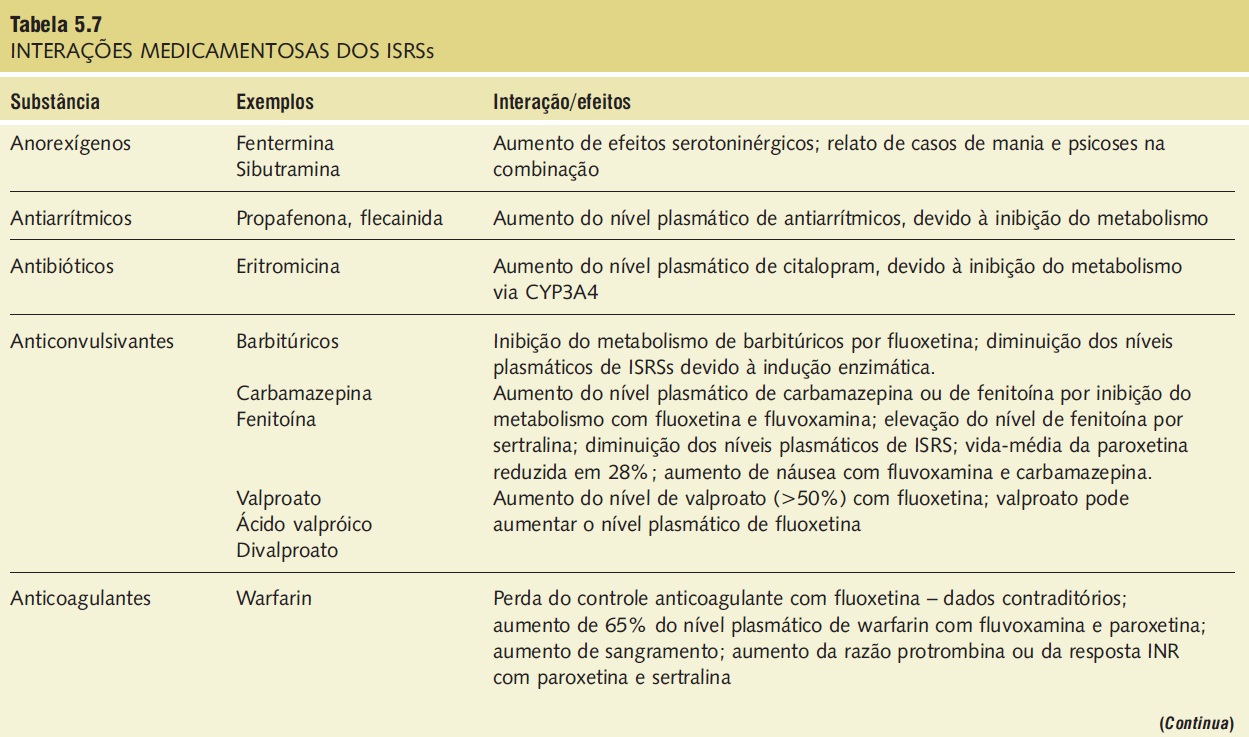
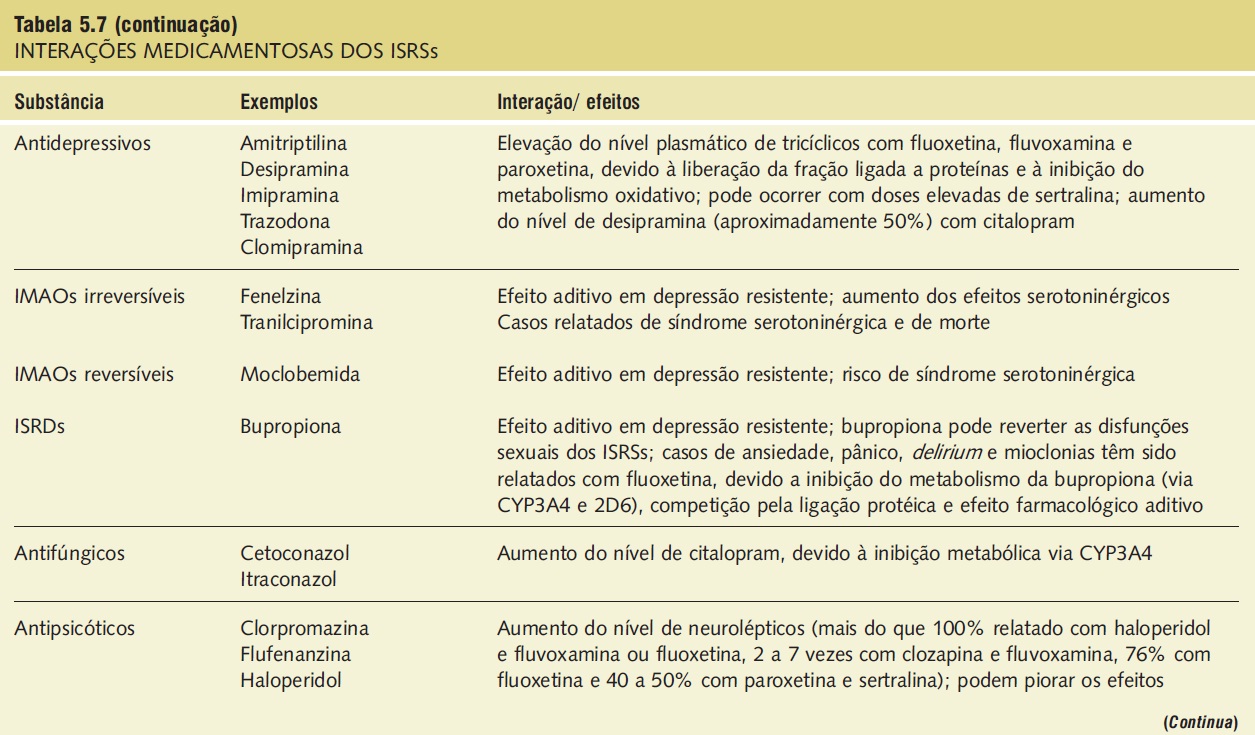
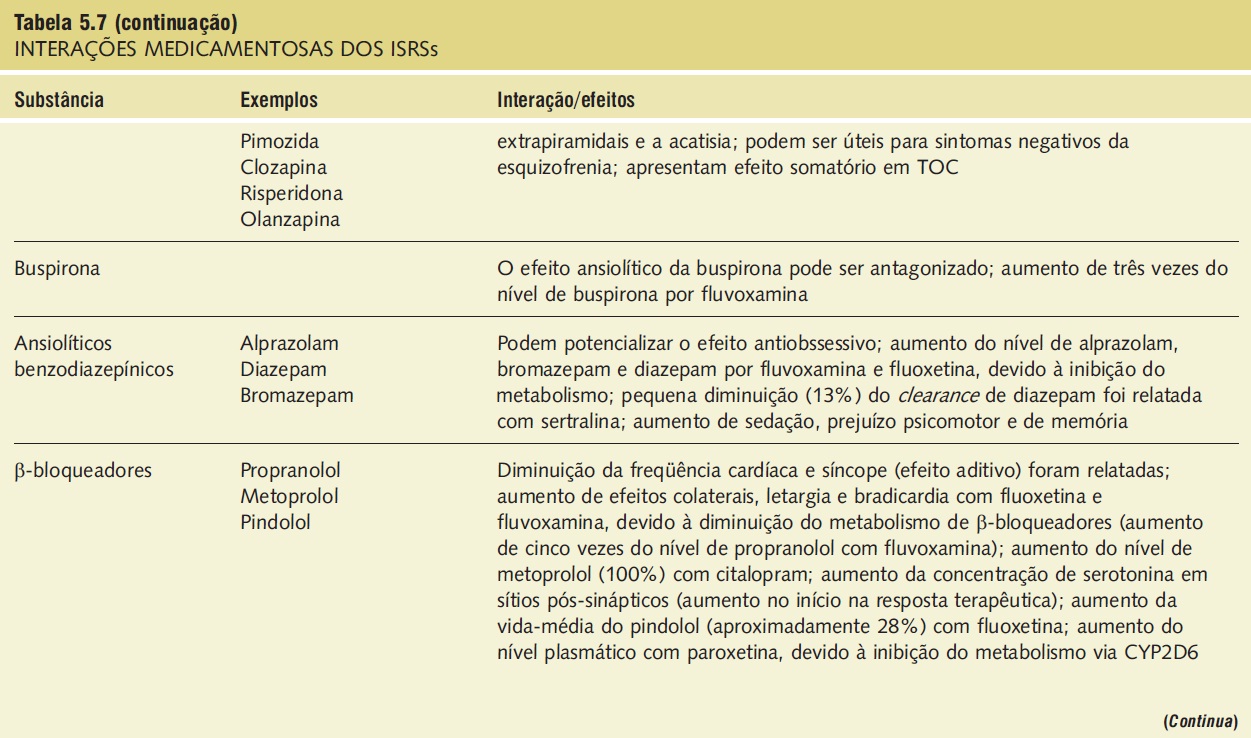
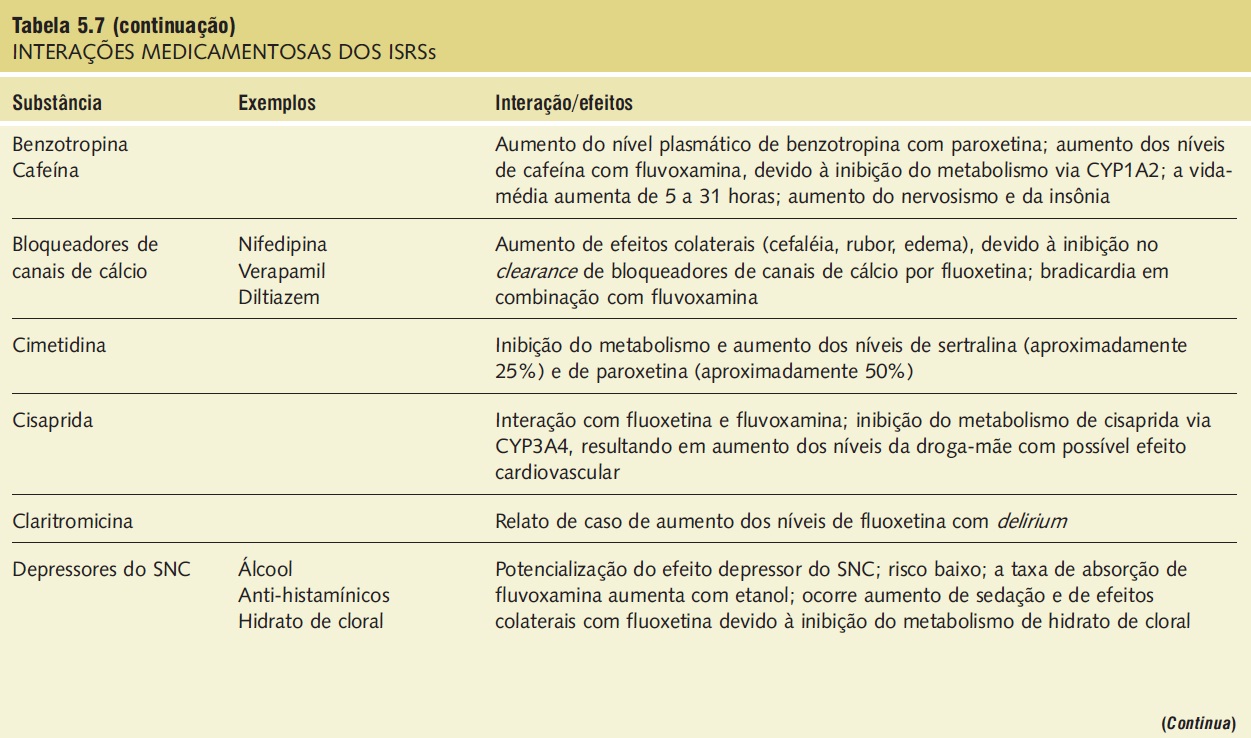
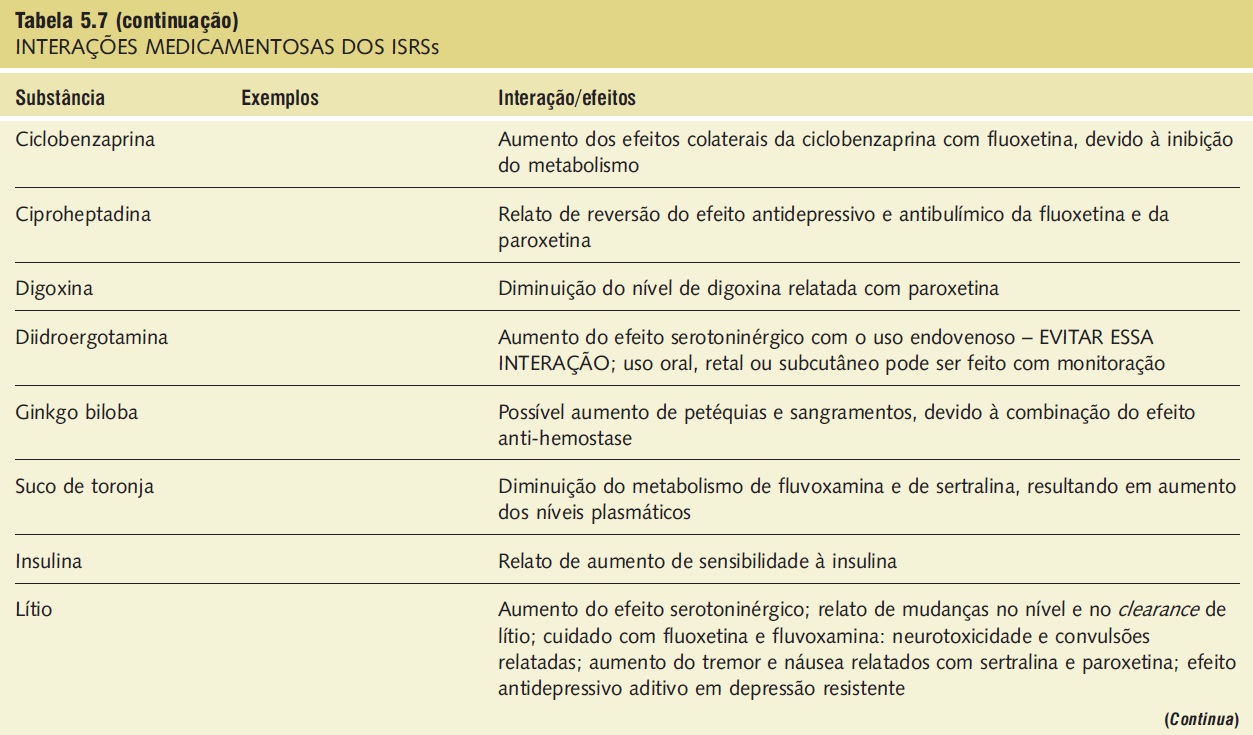
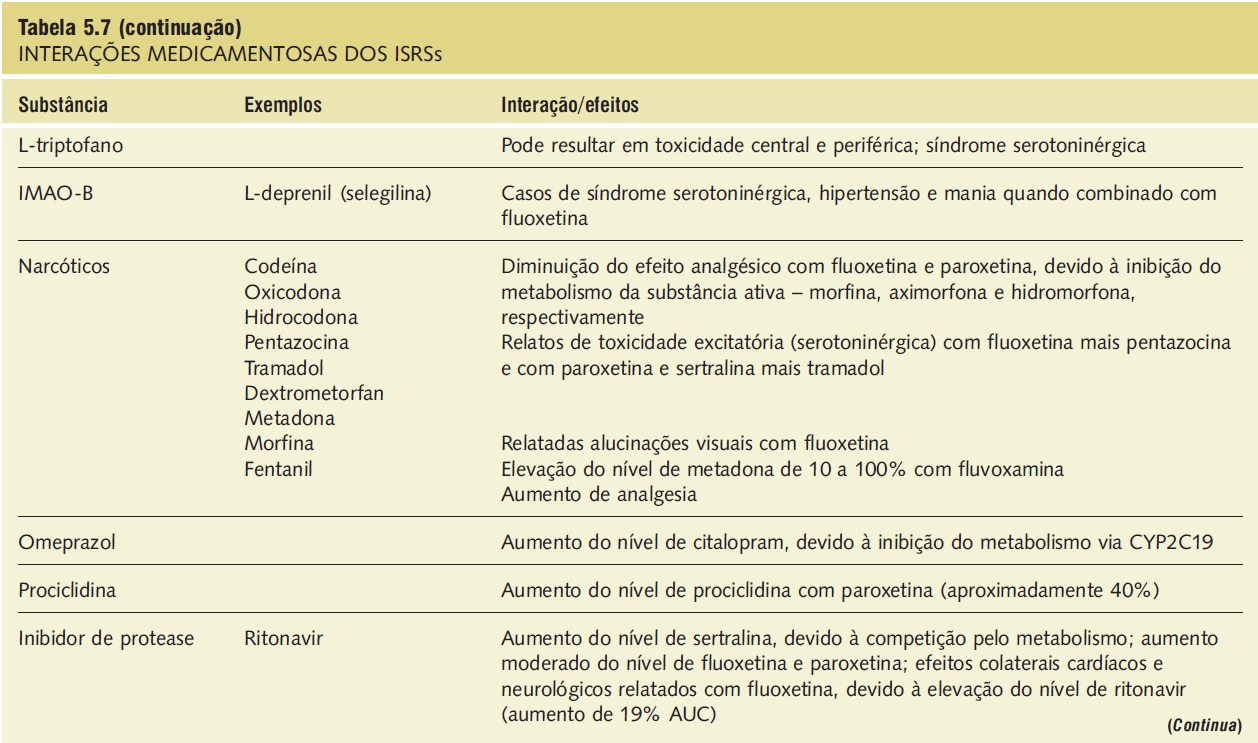
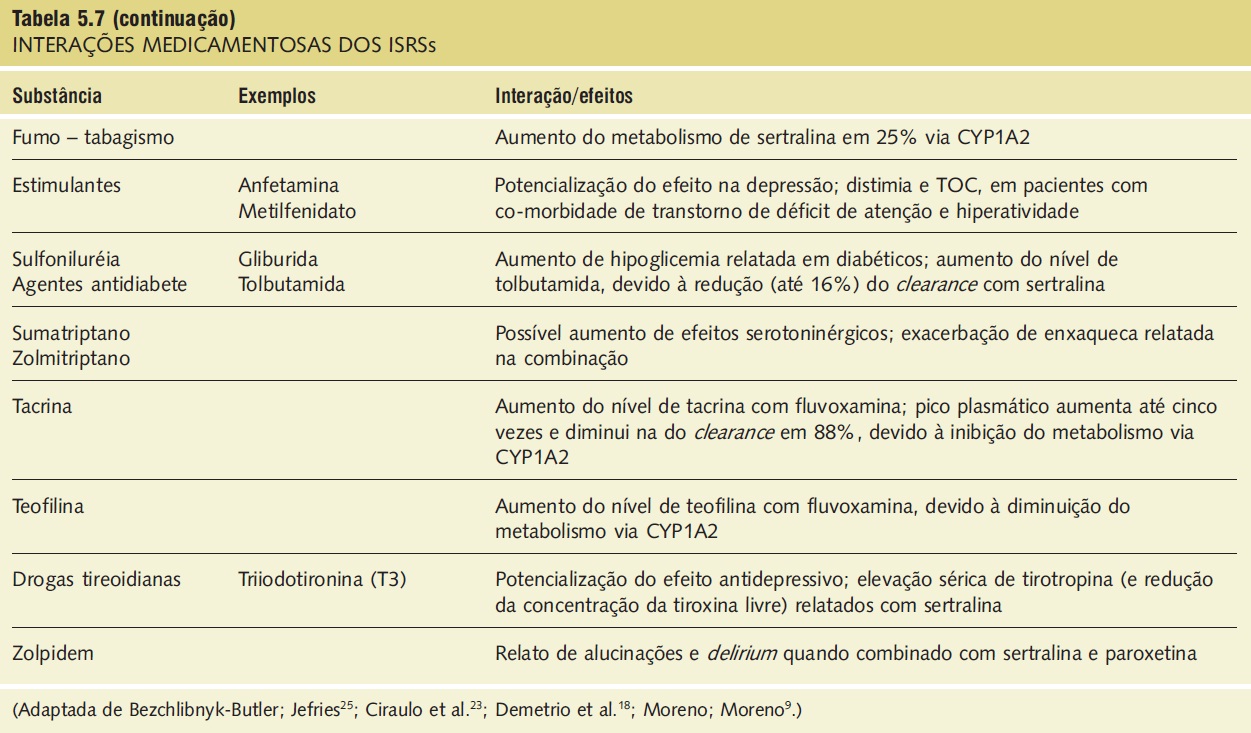
A síndrome serotoninérgica resulta do uso concomitante de substâncias serotoninérgicas (triptofano) e inibidores da monoaminoxidase. Outras substâncias implicadas incluem ADTs, trazodona, lítio, meperidina, buspirona e fenfluramina (Tab. 5.7).9,18,23,25 A síndrome ocorre em graus variados de gravidade e pode ser fatal. Os sinais e os sintomas podem ser confundidos com síndrome neuroléptica maligna. Sintomas sugestivos da síndrome incluem mudanças no estado mental do paciente (confusão, hipomania), agitação, mioclonias, hiper-reflexia, diaforeses, arrepios ou calafrios, tremor, diarreia, incoordenação e febre. O tratamento consiste na retirada das substâncias serotoninérgicas e no suporte e na observação da resolução espontânea do quadro (geralmente em 24 horas). Em casos graves, o tratamento com propranolol ou metisergida pode ser útil.
Não há evidências de teratogenicidade em humanos. Foi relatada maior incidência de partos prematuros com o uso de ISRSs no terceiro trimestre de gestação. Sintomas de abstinência em neonatos, tais como tremores, irritabilidade, inquietação e nervosismo puderam ser observados (no caso da fluoxetina, estão relacionados aos níveis plasmáticos dessa droga e da norfluoxetina). A fluoxetina e o citalopram são distribuídos para o leite materno em níveis terapêuticos, e seu uso em lactantes não é recomendado. O lactente pode receber até 17% da dose materna de fluoxetina. Com sertralina, paroxetina e fluvoxamina, as concentrações encontradas são muito pequenas. Entretanto, deve-se pesar individualmente a relação risco/benefício de seu uso. Para os demais ISRSs, não há estudos conclusivos em humanos.
Uso em hepatopatas: a redução na metabolização dos ISRSs em pacientes com comprometimento hepático pode implicar a necessidade de usar doses mais baixas. Pacientes com cirrose hepática alcoólica apresentam clearance reduzido de fluoxetina, de sertralina e de fluvoxamina.9,18,23,25
Assim como o perfil farmacocinético, o potencial para interações medicamentosas difere entre os vários ISRSs. O principal mecanismo das interações medicamentosas envolve a inibição de diferentes isoenzimas do citocromo P450: CYP2D6, CYP3A3/4, CYP1A2, CYP2C9/10 e CYP2C19.9,18,23,25
Tabela 5.7
Venlafaxina: É rapidamente absorvida, sua biodisponibinidade é de 45%, e a ingestão com alimentos retarda, mas não compromete sua absorção. A liberação da venlafaxina da formulação de liberação prolongada (XR) é controlada pela membrana celular e independe do pH. Embora a absorção da formulação de liberação prolongada ocorra mais lentamente e em concentrações plasmáticas inferiores, o total absorvido é o mesmo. A ligação proteica é moderada em cerca de 30%. A venlafaxina sofre metabolização hepática com importante efeito de primeira passagem. Estudos in vitro evidenciam o envolvimento do CYP3A4 na metabolização da venlafaxina.18,19,35
Milnaciprano: Apresenta boa absorção após administração por via oral. Sua biodisponibilidade é da ordem de 85%, não sendo modificada pela alimentação. A concentração plasmática máxima (Cmax) é atingida em aproximadamente duas horas (Tmax) após a ingestão oral. Essa concentração é da ordem de 120 ng/mL após uma ingestão única de 50 mg. O aumento da concentração plasmática é proporcional à dose até a concentração de 200 mg. Após ingestões repetidas, o nível plasmático do estado de equilíbrio é atingido em 2 a 3 dias. A variação individual é pequena. A taxa de ligação a proteínas plasmáticas é baixa (13%) e insaturável. Sua biotransformação é simples, limitando-se essencialmente a conjugação com ácido glicurônico. Não há metabólito ativo. O milnaciprano tem um clearance total da ordem de 40 L/h, e suameia-vida de eliminação plasmática é de aproximadamente oito horas. A eliminação é essencialmente por via urinária (90% da dose ingerida), com secreção tubular do produto na forma inalterada. Após ingestões repetidas, o milnaciprano é totalmente eliminado 2 a 3 dias após a interrupção do tratamento.13,18,19
Duloxetina: É bem absorvida depois de administrada por via oral, e sua concentração plasmática máxima (Cmax) ocorre seis horas após a administração. Quando ingerida com alimento, o pico de concentração é atingido em 6 a 10 horas, ocorrendo também uma discreta diminuição na absorção (aproximadamente 11%). Encontra-se altamente ligada (> 90%) às proteínas plasmáticas, principalmente à albumina e à glicoproteína a-1-ácida. A duloxetina é extensivamente metabolizada, e seus metabólitos são excretados principalmente na urina. As principais vias de biotransformação envolvem a oxidação do anel naftil, seguida por conjugação e posterior oxidação. Tanto CYP2D6 quanto CYP1A2 catalisam a formação dos dois principais metabólitos, o conjugado glucuronídeo da 4hidróxi duloxetina e o sulfato conjugado da 5-hidróxi,6-metóxi duloxetina. Os metabólitos circulantes não são farmacologicamente ativos. A meia-vida de eliminação é de 12 horas, e o clearance plasmático é de 101 L/h. A maior parte da duloxetina (70%) é recuperada na urina na forma de metabólitos, e aproximadamente 20% são recuperados nas fezes.13,18,19,36
Diferentemente dos ISRSs, o efeito de potente bloqueio serotoninérgico da venlafaxina é complementado por um efeito leve a moderado na recaptação da noradrenalina em dose de 150 mg. Em contraste, a duloxetina é um potente inibidor da recaptação de serotonina e noradrenalina em doses de 60 mg. Os ISRSNs promovem rápida downregulation de receptores ß-adrenérgicos acoplados à AMPc (adenosina monofostato cíclica). Esse efeito pode estar associado a um início de ação precoce. Em humanos, a dose terapêutica e as concentrações plasmáticas do milnaciprano produzem, consistentemente, um nível de inibição de 50 a 90% da recaptação de noradrenalina e serotonina.4
Venlafaxina: Provoca náuseas, tonturas e sonolência. Com doses acima de 225 mg/dia, podem aparecer sintomas como hipertensão, sudorese abundante e tremores. A hipertensão aparece como resultado da inibição da recaptação de noradrenalina, desenvolvendo-se em cerca de 3% dos pacientes que fazem uso de 100 mg/dia; em 5% dos pacientes em uso de doses entre 101 e 200 mg/dia; em 7% dos pacientes em uso de doses entre 201 e 300 mg/dia; e em 13% dos pacientes em uso de doses acima de 300 mg/dia. Porém, em menos de 1% dos pacientes, o tratamento deve ser interrompido por esse motivo. A magnitude do aumento nos níveis da pressão arterial é de 2 mm/Hg com doses de 225 mg/dia e de 7,5 mm/Hg com doses de 375 mg/dia. O tratamento da hipertensão, quando necessário, inclui o uso de drogas anti-hipertensivas. Os efeitos colaterais na esfera sexual aparentam ser dose-dependentes, e parece não haver desenvolvimento de tolerância. Podem ser relatados diminuição da libido, anorgasmia, retardo ejaculatório e impotência.35
Milnaciprano: Os efeitos colaterais mais comumente observados são disúria, palpitações, hipotensão, taquicardia, efeitos gastrintestinais (constipação, náusea, vômitos), boca seca, cefaléia, tremor, sudorese, tonteira e nervosismo. A disúria é mais frequente com milnaciprano do que com ADTs. Recomenda-se redução da dose em idosos e em pacientes com insuficiência renal, mas não em hepatopatas. Em pacientes com disfunção renal, o medicamento se concentra devido à redução significativa do seu clearance renal.
Duloxetina: Seus efeitos são constipação intestinal, boca seca, náusea, cefaleia, diarreia, vômito, diminuição do apetite, perda de peso, cansaço, tontura e sonolência. Ocorrem também tremores, sudorese, ondas de calor, visão borrada, anorgasmia, insônia, diminuição da libido, dificuldade de ereção e de ejaculação. Há relatos de dificuldade em urinar (apenas no sexo masculino), de diminuição do apetite, de fraqueza, de taquicardia, de tontura, de dilatação da pupila e de alterações visuais. Além desses, foram registrados efeitos de eructação, gastrenterite, estomatite, calafrios, sensação de calor e/ou frio, mal-estar, sede, aumento de peso, aumento da pressão sanguínea, alterações laboratoriais relacionadas à função hepática, desidratação, rigidez e contração musculares, alteração do paladar, ansiedade, distúrbio do sono, agitação, bruxismo, desorientação, além de aumento da frequência urinária noturna, bocejo, suores noturnos, reação de sensibilidade à luz, rubor facial e extremidades frias.
Venlafaxina18,19: Não há estudos controlados em gestantes. Estudos com ratos e coelhos, empregando doses superiores às terapêuticas em até 12 vezes, não evidenciaram efeitos teratogênicos. Não se sabe se a venlafaxina é excretada no leite materno, porém não foram relatados problemas a esse respeito. O uso em pacientes com doenças cardiovasculares e hipertensão deve ser bem avaliado, uma vez que a indução de elevação nos níveis da pressão arterial ou a hipotensão postural podem agravar condições pré-existentes. O metabolismo da venlafaxina está alterado em pacientes com comprometimento hepático, e deve-se considerar redução nas doses em até 50% no caso de comprometimento hepático grave ou moderado. A excreção da venlafaxina pode ser alterada em pacientes com comprometimento renal. Pacientes com comprometimento leve ou moderado devem receber 25 a 50% da dose. Pacientes em hemodiálise devem receber 50% da dose, que deve ser administrada após a sessão de diálise.
Milnaciprano18,19: É conhecida a hipersensibilidade ao milnaciprano ou a outros componentes da formulação. É contraindicado em pacientes com menos de 15 anos. Também é desaconselhado seu uso, na falta de dados clínicos, em associação com inibidores não seletivos da MAO (IMAOs), com inibidores seletivos da MAOB, digitálicos e agonistas 5HT1D (sumatriptano). Seu uso é contraindicado em casos de hipertrofia prostática e outras disfunções geniturinárias. Embora a interação com álcool não tenha sido evidenciada, recomenda-se evitar a ingestão de álcool, como com qualquer medicamento psicotrópico. Um aumento maior da medicação pode ocorrer em idosos e em pacientes com insuficiência renal.
O milnaciprano deve ser prescrito com prudência nos seguintes casos:
Ø pacientes com insuficiência renal: a posologia deve ser reduzida, em razão do prolongamento da meia-vida de eliminação;
Ø pacientes com história de dificuldade para a passagem da urina, notadamente em pacientes com hipertrofia prostática e outros distúrbios geniturinários: devido ao componente noradrenérgico do mecanismo de ação do milnaciprano, é necessária a monitoração de doenças miccionais;
Ø pacientes hipertensos ou cardiopatas: recomenda-se reforçar a vigilância clínica, uma vez que o milnaciprano pode aumentar, discretamente, a frequência cardíaca em alguns pacientes;
Ø pacientes com glaucoma de ângulo estreito;
Ø pacientes com epilepsia ou história de epilepsia:esse medicamento deve ser usado com cuidado e descontinuado em todos os casos em que ocorrerem convulsões.
Existem relatos de casos de hiponatremia em pacientes recebendo inibidores da recaptação de serotonina, possivelmente devido a uma síndrome de secreção inadequada do hormônio antidiurético. Cuidados especiais são recomendados com idosos, com pacientes em uso de diuréticos, ou outros medicamentos que possam induzir hiponatremia, e com pacientes cirróticos ou desnutridos.
Com relação à gravidez e à lactação, por medida de precaução, é preferível não usar o milnaciprano durante a gravidez, pois não existem informações suficientes sobre seus efeitos sobre o feto; como o milnaciprano passa para o leite em pequenas quantidades, seu uso é contraindicado durante a amamentação.
Estudos em animais evidenciaram a passagem de pequena quantidade de milnaciprano pela placenta. Atualmente, não há dados relevantes que demonstrem efeitos teratogênicos ou tóxicos desse medicamento para o feto quando administrado durante a gravidez. Na ausência de efeitos teratogênicos demonstrados nos estudos em animais, más formações em humanos não são esperadas. Entretanto, por medida de precaução, é preferível, como dissemos, não administrar o milnaciprano durante a gravidez.
Duloxetina18,19: Não houve estudos bem controlados e adequados em gestantes. Devido ao fato de os estudos em animais nem sempre predizerem a resposta em humanos, esse medicamento deve ser usado em gestantes somente quando o benefício potencial justificar o risco para o feto.
Deve-se ter cuidado especial com relação à sua administração em idosos (embora não seja necessário ajuste de dose), em pacientes com transtorno bipolar, com história prévia de mania, com convulsão, com aumento de pressão intraocular, com doença cardíaca e com hipertensão. Em pacientes com risco de hemorragia ou em uso de anticoagulantes e de antiagregantes plaquetários, deve-se manter a atenção. Não se deve usar esse medicamento em crianças ou em pacientes com menos de 18 anos. Podem ocorrer diminuição da função hepática, diminuição grave da função renal e hipertensão não controlada. Deve-se atentar aos pacientes que fizeram uso de IMAO nos 14 dias anteriores ao uso de duloxetina, bem como aos pacientes em uso de fluvoxamina, de ciprofloxacina ou de enoxacina, aos que têm intolerância à frutose ou àqueles com má absorção de glicose-galactose e com insuficiência desucrose-isomaltase (cápsulas de duloxetina contêm sucrose).
Os ISRSNs podem precipitar síndrome serotoninérgica quando combinados com IMAOs, e, portanto, essa combinação é contraindicada. É recomendável aguardar duas semanas após a interrupção do IMAO para iniciar o ISRSN. Em função de a venlafaxina apresentar uma meia-vida curta (cinco horas para venlafaxina e 11 horas para seu principal metabólito, O-desmetilvenlafaxina), uma semana de wash-out é suficiente antes de iniciar o IMAO. Venlafaxina e duloxetina são inibidores fracos do citocromo 2D6. À semelhança de citalopram e escitalopram, os ISRSNs não possuem efeito potente de inibição de outras enzimas hepáticas.
Entretanto, ambos são metabolizados pela enzima 2D6 e, de alguma forma, pela enzima 1A2. Portanto, cimetidina, paroxetina e outros medicamentos que inibem 2D6 podem resultar em aumento pronunciado da pressão arterial e em outros efeitos colaterais. Venlafaxina pode aumentar os níveis de haloperidol, mas esse efeito não é mediado pelas enzimas 2D6 ou 1A2. As interações medicamentosas de significado clínico estão descritas na Tabela 5.8.
É um derivado da triazolopiridina, sintetizado na Itália em 1966 e disponível nos Estados Unidos apenas no início dos anos 1980. Foi considerada um dos primeiros antidepressivos de segunda geração; algumas vezes é também chamada de atípica, possuindo propriedades farmacológicas e bioquímicas muito específicas e não totalmente compreendidas, o que continu a estimular novas pesquisas.
Farmacocinética. A trazodona é bem absorvida pelo trato gastrintestinal e, se ingerida às refeições, ou imediatamente após, pode haver aumento na quantidade absorvida, redução na concentração máxima e aumento no tempo necessário para atingir pico plasmático. Em geral, os picos plasmáticos são atingidos em duas horas. Apresenta alta ligação proteica de cerca de 90% e sofre hidroxilação hepática. Sua meia-vida é de 6 a 11 horas. A eliminação é principalmente renal (75%, predominantemente como metabólitos inativos) e biliar (20%).
Mecanismo de ação. O mecanismo de ação postulado para a trazodona envolve a inibição da recaptação de serotonina e noradrenalina. Em longo prazo, ocorrem a dessensibilização e a diminuição no número de receptores ß-adrenérgicos e 5HT2A. Esse medicamento apresenta atividade antagonista de receptores a-1-adrenérgicos e anti-histamínicos, que estão mais relacionados aos seus efeitos colaterais. O metabólito ativo mCPP também apresenta algum grau de atividade serotoninérgica pós-sináptica.
Populações especiais e precauções. O uso da trazodona na gestação está contraindicado, pois estudos em animais associam seu uso a más-formações fetais. A trazodona é excretada no leite materno e, por isso, o aleitamento é contraindicado quando a lactante faz uso dessa droga. Em pacientes com comprometimento hepático e/ou renal, seu uso deve ser feito com cautela, em função de alterações no metabolismo e na excreção da droga.
Efeitos colaterais. Os efeitos colaterais mais frequentes da trazodona são sedação, hipotensão ortostática, tonturas, cefaléia, náuseas e boca seca. Reações alérgicas e irritação gástrica podem parecer. Alguns relatos de casos sugerem associação entre a trazodona e o aparecimento de arritmias em pacientes que já apresentavam contrações ventriculares prematuras ou prolapso de válvula mitral. A trazodona está associada à ocorrência de priapismo (ereção peniana prolongada na ausência de estímulo). Nesse caso, deve-se suspender o uso do medicamento. Sugere-se avaliar com o paciente a troca do antidepressivo caso se perceba que a frequência e a duração das ereções estão aumentando. O tratamento do priapismo consiste na injeção intracavernosa de solução de epinefrina (1 mcg/mL). Outras disfunções sexuais também podem aparecer.31
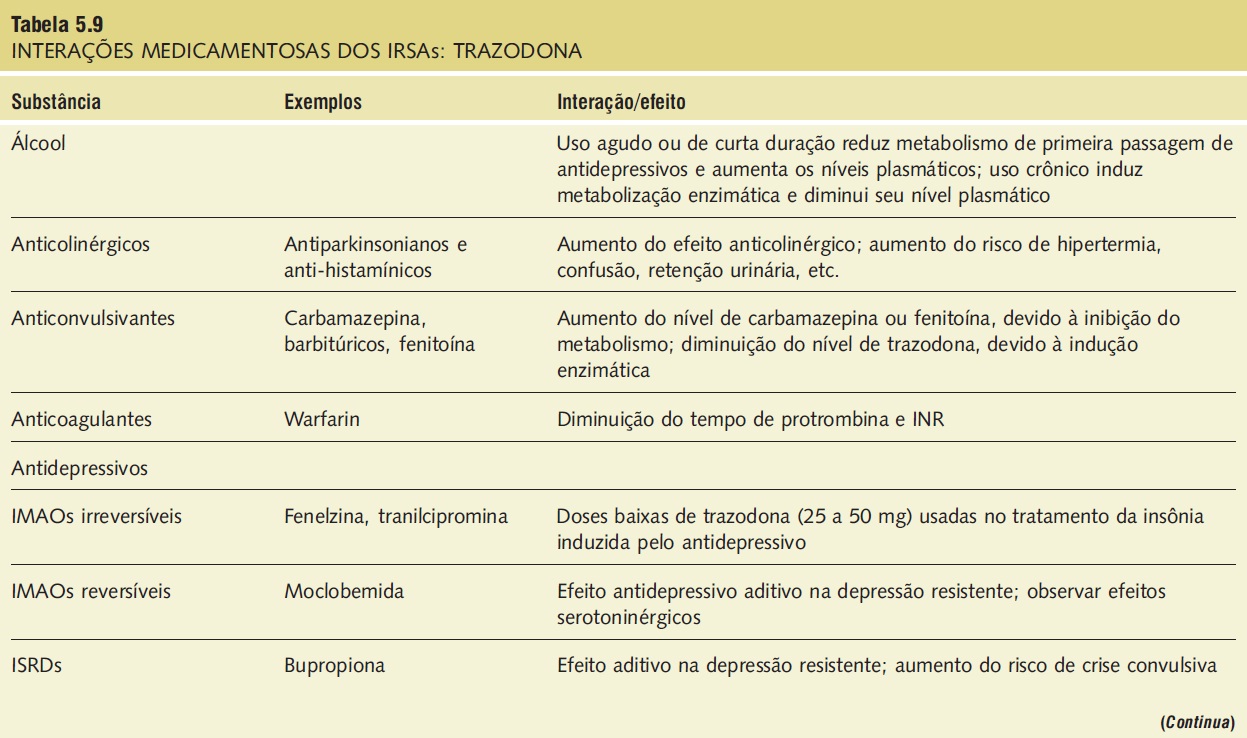
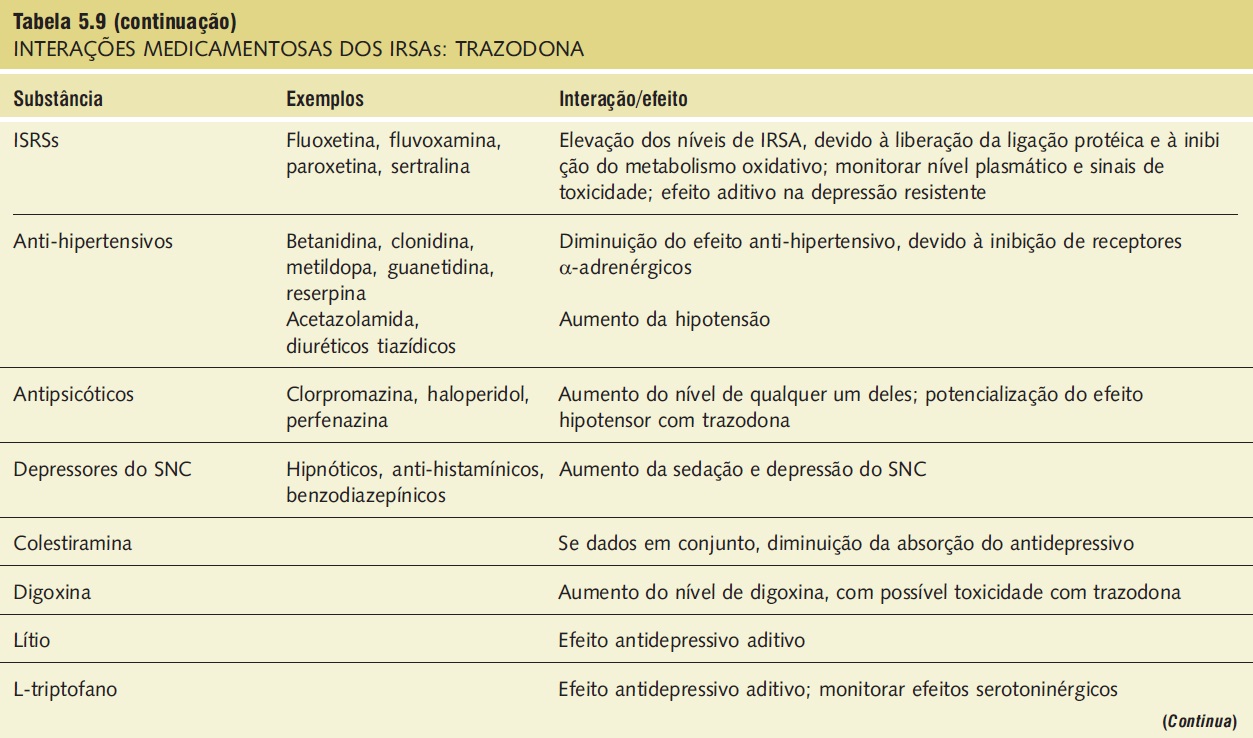
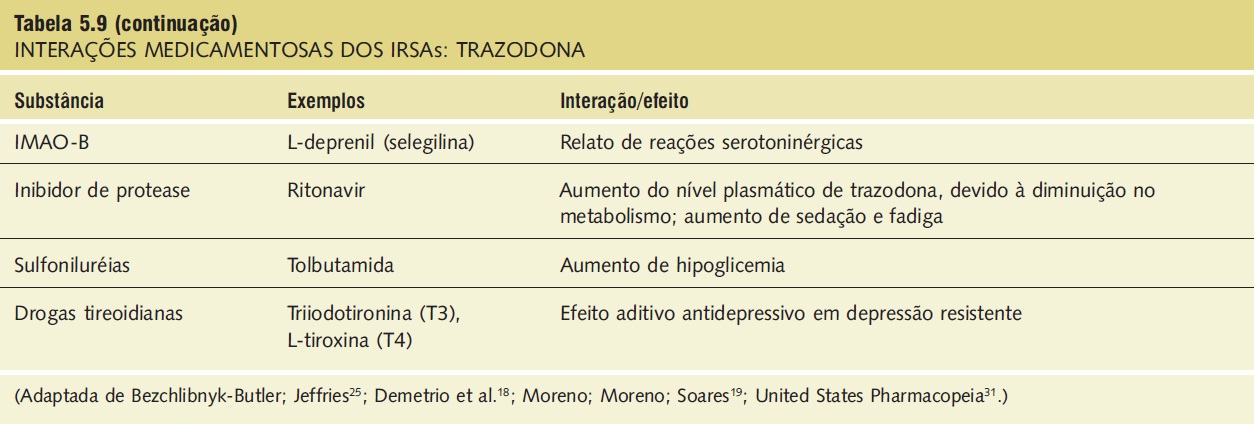
Interações medicamentosas. As interações medicamentosas clinicamente significativas envolvem substâncias depressoras do SNC e IMAOs e estão descritas na Tabela 5.9.
Intoxicação. Casos de intoxicação por trazodona frequentemente se caracterizam por apresentarem sedação, hipotensão, perda de coordenação muscular, náuseas e vômitos. O tratamento consiste na redução da absorção com lavagem gástrica e administração de carvão ativado, na tentativa de aumento da eliminação com diurese forçada e na adoção de medidas de monitoração cardíaca e de suporte.24
Tabela 5.8
Tabela 5.9
A reboxetina foi o primeiro composto de uma nova classe de antidepressivos (inibidores da recaptação de noradrenalina – ISRNs), estruturalmente semelhantes à viloxazina, a ser comercializado. Apresenta atividade seletiva sobre a recaptação de noradrenalina, com atividade antagonista a-2. Não possui efeitos significativos sobre receptores colinérgicos, histamínicos e a-1-adrenérgicos, ou na inibição da monoaminoxidase, e seu efeito antidepressivo foi descrito inicialmente na década de 1980.
Farmacocinética. A reboxetina é absorvida pelo trato gastrintestinal e apresenta alta ligação à glicoproteína a-1 plasmática. Atinge pico plasmático em 1,5 a 2,5 horas. Sofre metabolização hepática por hidroxilação e por oxidação, principalmente. Não interage com isoenzimas do sistema citocromo P450. Sua meia-vida é de 12 a 13 horas, e sua eliminação se dá pela urina (76% na forma inalterada e em metabólitos) e pelas fezes (7 a 16%).33
Populações especiais e precauções. Quanto ao uso na gestação e na lactação, ainda não há dados. Em pacientes com doenças cardiovasculares, a reboxetina pode levar a aumento de frequência cardíaca e a um leve decréscimo na pressão arterial. Nesse caso, o medicamento deve ser usado com precaução. Em pacientes idosos, não há estudos. Entretanto, pacientes com hipertrofia prostática podem sentir-se especialmente incomodados pela retenção urinária. A meia-vida prolongada requer ajuste de doses, ainda sem diretrizes definidas. A extensa metabolização hepática sofrida pela reboxetina sugere a necessidade de ajustes de doses em pacientes hepatopatas, porém nesse caso também não há condutas definidas.20
Efeitos colaterais. Os efeitos colaterais mais significativos da reboxetina são taquicardia, impotência, hesitação ou retenção urinária, insônia, sudorese excessiva, obstipação intestinal e boca seca. Em geral, esses efeitos têm intensidade moderada, mesmo em doses acima de 8 mg/dia.20,31,33,37
Interações medicamentosas. A ausência de interação com as enzimas do citocromo P450 e a seletividade da ação conferem à reboxetina baixo potencial de interações medicamentosas. Embora ainda não existam estudos definitivos sobre o assunto, os já realizados demonstraram que a reboxetina não apresenta interação farmacocinética com o lorazepam em indivíduos saudáveis, e também não se observou interação farmacodinâmica (desempenho psicomotor ou cognitivo) com o álcool em estudo duplo-cego, que incluiu número reduzido de pacientes.
A bupropiona é um antidepressivo unicíclico, que teve sua comercialização retardada devido a pendências acerca do risco de induzir convulsão, mas foi liberada quando ficou claro que esse risco é dose-dependente e tende a ocorrer em populações específicas. Em 1998, foi comercializada a formulação SR, de liberação sustentada, podendo ser administrada duas vezes ao dia e com menor risco de convulsão. Já em 2003, a formulação XL aparece para ser administrada uma vez ao dia.
Farmacocinética. A bupropiona é rapidamente absorvida pelo trato intestinal, porém o metabolismo pré-sistêmico elevado diminui sua biodisponibilidade. Esse medicamento e a hidroxibupropiona apresentam alta ligação proteica (84 e 77%, respectivamente). A bupropiona cruza rapidamente a barreira hematoencefálica e a placenta, sendo distribuída no leite materno. É extensivamente metabolizada (inclusive com metabolização pré-sistêmica), e três de seus metabólitos apresentam alguma atividade, segundo estudos em animais: a hidroxibupropiona (formada principalmente pelo citocromo P450 2B6), com potência equivalente à bupropiona, a treoidrobupropiona e a eritroidrobupropiona, que são formadas por hidroxilação e/ou redução e apresentam de um décimo à metade da potência da bupropiona. Suameia-vida de distribuição é de cerca de 3 a 4 horas, e a meiavida de eliminação após dose única é de 14 horas; no estado de equilíbrio, a meia-vida é de 21 horas (podendo variar entre 12 e 30 horas). A meia-vida de eliminação da hidroxibupropiona é de cerca de 20 horas. Os picos plasmáticos da bupropiona e da hidroxibupropiona são de 1,5 e 3 horas, respectivamente, passando para o dobro quando em uso da formulação de liberação prolongada. A eliminação renal é de 1% na forma inalterada; como metabólitos, acima de 60% em 24 horas e acima de 80% em 96 horas. A eliminação fecal é de 10%, principalmente na forma de metabólitos.31
Mecanismo de ação. Embora não seja completamente conhecido, o mecanismo de ação da bupropiona se dá por meio de suas atividades noradrenérgica e dopaminérgica. A bupropiona aumenta a liberação de noradrenalina corpórea, e é um fraco inibidor in vitro da captação neuronal de noradrenalina e de dopamina, porém de relevância farmacológica. Sua ação inibitória da recaptação de dopamina é menor do que a observada com sertralina. Entretanto, sua ação dopaminérgica é importante devido ao fato de os níveis de ácido homovanílico, o metabólito primário da dopamina, diminuírem em pacientes que respondem à bupropiona, mas não naqueles que não respondem. Nos últimos anos, sua ação noradrenérgica ficou mais evidente. A hidroxibupropiona é seu metabólito ativo. A bupropiona não inibe a monoaminoxidase e tem pouca afinidade pelo sistema serotoninérgico. Também não interage com receptores histamínicos e colinérgicos, permitindo maior tolerabilidade.26,35
Populações especiais e precauções. Estudos adequados e bem controlados em humanos ainda não foram realizados, porém sabe-se que a bupropiona passa rapidamente à placenta. Seu uso na gestação não é recomendado.24 A bupropiona é distribuída para o leite materno, oferecendo risco potencial, como convulsões, para o lactente. Em pacientes geriátricos acima de 60 anos, não há limitações ao uso de bupropiona. No entanto, alterações metabólicas relacionadas à idade podem causar intolerância aos efeitos colaterais, e alterações renais e hepáticas podem exigir redução nas doses prescritas. Em pacientes com história de traumatismo craniano, tumores cerebrais, quadros cerebrais orgânicos ou alterações eletroencefalográficas, o uso da bupropiona não é recomendado, em função do risco de convulsões. Em doentes renais ou hepatopatas, a metabolização e a excreção da bupropiona podem ficar alteradas. Sugere-se iniciar o tratamento com baixas doses e monitorar intensivamente o paciente. Na anorexia e na bulimia, não é recomendado o uso dessa droga, pelo maior risco de convulsões.31
Efeitos colaterais. A bupropiona apresenta boa tolerabilidade. Entre os antidepressivos de nova geração, apresenta o menor potencial de indução de efeitos colaterais e a menor incidência de descontinuação do tratamento por intolerância.
Os efeitos colaterais mais frequentemente observados são agitação, ansiedade, rash cutâneo, diminuição do apetite, boca seca e obstipação intestinal. Entretanto, o risco de indução de convulsões é maior do que o de outros antidepressivos, e mais frequente com doses elevadas. A incidência de convulsões com a forma de liberação prolongada é de 0,1% com doses de até 300 mg/dia e de 0,4% com doses acima de 400 mg/dia. Com o uso da forma de liberação imediata, o risco passa para 0,4% com doses entre 300 e 450 mg/dia, podendo aumentar até 10 vezes em doses entre 450 e 600 mg/dia.38 Para minimizar o risco de convulsões, recomenda-se que cada dose do composto de liberação imediata não exceda 150 mg, e que do composto de liberação prolongada a dose não exceda 200 mg. Deve-se observar intervalo de quatro horas entre as doses do composto de liberação imediata, e de oito horas entre as tomadas do composto de liberação prolongada.
Intoxicação. Os efeitos clínicos da ingestão de doses elevadas de bupropiona são alucinações, diminuição do nível de consciência, náuseas, vômitos, convulsões (em um terço dos casos) e taquicardia, que pode evoluir para bradicardia e assistolia. O tratamento da intoxicação inclui medidas para diminuição da absorção. Pacientes estuporosos ou comatosos devem ser entubados. Em seguida, deve-se realizar lavagem gástrica e administrar carvão ativado a cada seis horas, se a ingestão ocorreu nas 12 horas anteriores. Não se recomenda o xarope de ipeca para induzir vômitos pelo risco de convulsões. No caso de convulsões, deve-se administrar benzodiazepínicos por via endovenosa. É fundamental monitorar o eletrocardiograma e o eletroencefalograma por pelo menos 48 horas, bem como o equilíbrio eletrolítico e ácido-básico em pacientes com estado de mal epiléptico. Medidas gerais de suporte, como diurese forçada, diálise ou hemoperfusão, não são indicadas, pois a bupropiona e seus metabólitos apresentam lenta difusão dos tecidos para o plasma.
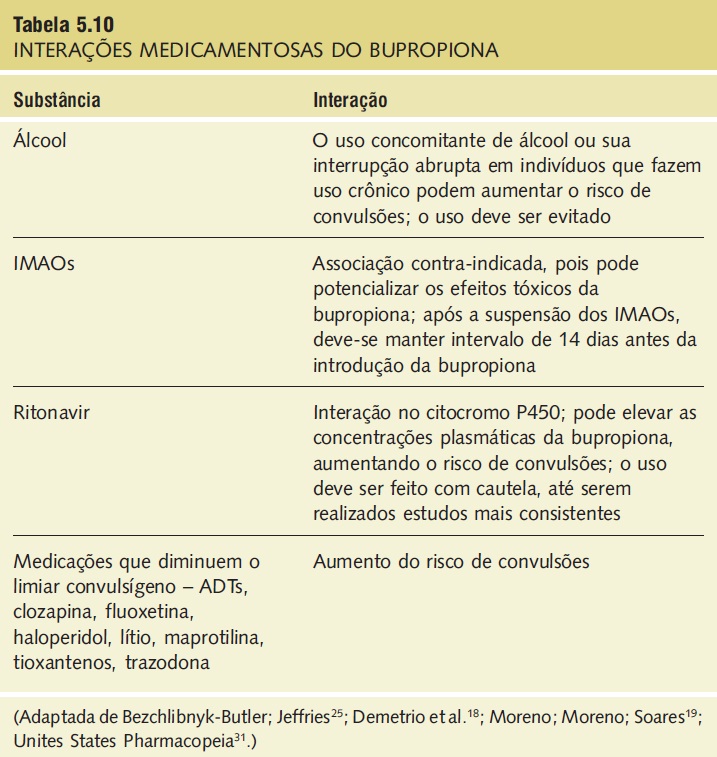
Interações medicamentosas. As interações medicamentosas significativas estão descritas na Tabela 5.10.
Tabela 5.10
A mirtazapina é clinicamente relacionada com a mianserina, um medicamento largamente utilizado durante anos na Europa. Embora tenha sido aprovada para o tratamento da depressão, possui outras indicações.
Mecanismo de ação. A ação da mirtazapina se dá pelo aumento das atividades noradrenérgica e serotoninérgica centrais. A mirtazapina é um antagonista de auto e heterorreceptores a-2 adrenérgicos pré-sinápticose antagonista 5-HT2 e 5-HT3 pós-sináptico. Apresenta fraca afinidade pelos receptores 5-HT1A e pelos 5-HT1B pós-sinápticos. Sua afinidade pelos receptores histamínicos H1 explica seu efeito sedativo. Apresenta fraca afinidade por receptores muscarínicos e dopaminérgicos.12,26
Farmacocinética. A mirtazapina é bem absorvida pelo trato gastrintestinal. Porém, devido ao metabolismo de primeira passagem, sua biodisponibilidade é de 50%. Apresenta alta ligação a proteínas plasmáticas (85%). Os picos plasmáticos são atingidos em cerca de duas horas, e o estado de equilíbrio em cinco dias, apresentando relação linear com a dose ingerida. A mirtazapina sofre metabolização hepática, principalmente desmetilação e hidroxilação, seguida de conjugação ao ácido glicurônico. Seus metabólitos são ativos e encontrados em níveis baixos. A meia-vidade eliminação é de 20 a 40 horas (mais longa em mulheres de todas as idades). Os metabólitos são eliminados na urina (75%) e nas fezes (15%).31
Populações especiais e precauções. Sugere-se evitar o uso de mirtazapina na gestação, e não se sabe se ela passa para o leite materno. A diminuição da eliminação da mirtazapina em idosos (40% entre homens e 10% entre mulheres) e a diminuição da função renal podem exigir ajustes de dose. Recomenda-se iniciar a administração com 7,5 mg ao dia e aumentar a dose para 15 mg em uma a duas semanas, dependendo da resposta do paciente e dos efeitos colaterais, devendo-se monitorar a sedação e o efeito anticolinérgico. Em indivíduos sem comprometimento hepático, sob o uso de mirtazapina, foram observadas elevações no número de transaminases hepáticas, superando em três vezes seu valor normal, sem o desenvolvimento de sinais e de sintomas de alteração da função hepática. Esses valores retornaram aos parâmetros normais com a suspensão da droga. Pacientes com comprometimento da função hepática apresentam diminuição de 30% do clearance após ingestão de dose única de 15 mg.31
Efeitos colaterais. A mirtazapina apresenta boa tolerabilidade. Os efeitos colaterais mais frequentemente relatados são sedação excessiva, ganho de peso (principalmente com o uso de doses baixas), boca seca, edema, obstipação intestinal e dispneia. Em estudos clínicos realizados antes de seu lançamento, observou-se a ocorrência de dois casos (entre 2.796 pacientes) de agranulocitose reversível e um caso de neutropenia grave, também reversível.12,31 Sugere-se que a mirtazapina seja suspensa em pacientes que apresentarem febre ou outros sinais de infecção e tiverem baixa contagem de leucócitos.31
Intoxicação. A mirtazapina apresenta alguma segurança em casos de intoxicação (relato de ingestão de até 30 vezes a dose recomendada), sendo mais segura do que a imipramina. Os sinais e os sintomas presentes em casos de intoxicação por mirtazapina incluem desorientação, tonturas, comprometimento de memória, taquicardia e sedação excessiva.12,31
O tratamento inclui medidas de suporte geral e monitoração das funções vitais. Podem-se empregar medidas para reduzir a absorção, como indução de emese e lavagem gástrica seguida da administração de carvão ativado.
Interações medicamentosas. As principais interações medicamentosas da mirtazapina estão sintetizadas na Tabela 5.11.
Tabela 5.11
Flutuações circadianas na temperatura corporal, secreções endócrinas e outros parâmetros biológicos, desorganização dos ritmos internos afetando fase, período ou amplitude têm sido efeitos comumente descritos nos pacientes com depressão.
Isso sugeria que novos tratamentos direcionados a tais alterações teriam um potencial valor no manejo da depressão. Devido ao fato de a melatonina ser conhecida como uma chave neuro-hormonal para a sincronização de ritmos biológicos, possíveis alterações em sua secreção foram investigadas em pacientes depressivos. Troca de período e mudanças na amplitude do ritmo da secreção de melatonina foram associadas à depressão. Consequentemente, levantou-se a interessante possibilidade de que um medicamento capaz de mimetizar os efeitos da melatonina e que facilmente atravessasse a barreira hematoencefálica seria benéfico para ressincronizar os ritmos alterados e para exercer efeito antidepressivo no caso de haver relação entre tais alterações e depressão. Essa hipótese levou à síntese da agomelatina, um agonista com alta afinidade pelos receptores clonados MT1 e MT2 de melatonina humana e que também demonstra propriedades antagonistas nos receptores 5-HT2c.39,40
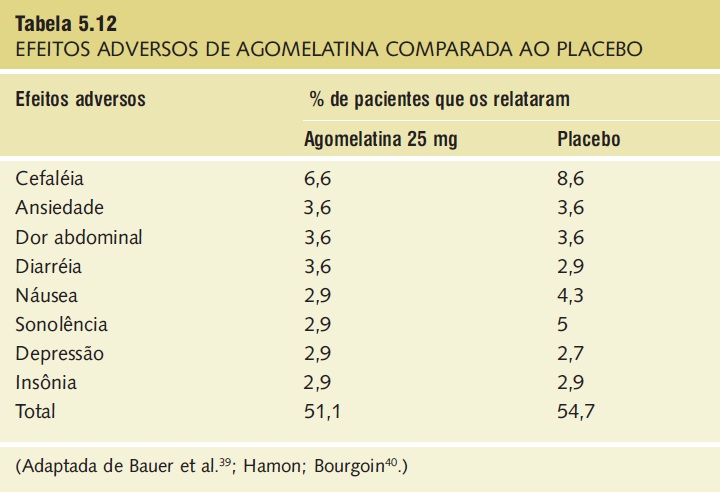
A agomelatina demonstrou ter um perfil favorável de efeitos adversos. Os mais comuns foram cefaleia, náusea e fadiga (Tab. 5.12).
O avanço da pesquisa em psicofarmacologia de antidepressivos vem oferecendo aos pacientes substâncias com perfis farmacocinéticos, com tolerância e com interações com outras drogas bastante diferentes entre si. Apesar disso, os mecanismos de ação propostos para cada uma delas permanecem vinculados às teorias de monoaminérgicos, de aumento da oferta de neurotransmissores na fenda sináptica e de subsensibilização de receptores pós-sinápticos. A seletividade de ação não constitui fator único para a ação dos antidepressivos, porque, a partir da ligação da substância com o receptor, várias vias intracelulares podem ser seguidas, o que desfaz sua “ação seletiva”. Tratamentos com antidepressivos, além de aumentarem os níveis sinápticos de noradrenalina e serotonina, promovem a ativação de cascatas de sinais de transdução intracelulares, uma das quais é a cascata do monofosfato de adenosina cíclico – transcription factor cAMP response element binding protein (AMPc-CREB), que tem como gene alvo o brain derived neurotrophic factor (BDNF), fator conhecido por exercer uma forte influência no desenvolvimento, na sobrevivência, na manutenção e na plasticidade neuronal dentro do sistema nervoso, tanto imaturo quanto adulto.41,42 A partir dessas evidências, postula-se a possível ação neurotrófica dos antidepressivos em pacientes deprimidos. Em adição ao papel da ação neurotrófica na sobrevivência celular, é possível que os antidepressivos também regulem outros processos, como a neurogênese. Estudos sustentam essa hipótese e demonstram que o tratamento crônico aumenta a neurogênese, por exemplo, nas células do giro denteado.29
Comparando os novos antidepressivos aos clássicos ADTs e IMAOs, verificase um esforço no sentido de aperfeiçoar cada vez mais a ação em sítios receptores determinantes da eficácia clínica, evitando aqueles responsáveis pelos efeitos colaterais. Do amplo espectro de ação dos antidepressivos clássicos, passou-se aos ISRSs, mais bem tolerados e mais seguros na superdosagem, cuja ação é praticamente restrita à inibição da recaptação da serotonina. Apesar de os estudos existentes igualarem sua eficácia à dos ADTs, permanecem dúvidas em relação à resposta terapêutica em deprimidos graves. Diferenças em sua farmacocinética e no seu potencial de interações medicamentosas tornam o grupo heterogêneo, passível de indicações em diferentes situações clínicas.
Os novos antidepressivos buscaram aliar o amplo espectro de ação dos antidepressivos clássicos à tolerabilidade e à segurança dos ISRSs. Assim, surgiram moclobemida, trazodona, nefazodona, bupropiona, reboxetina, mirtazapina, venlafaxina, duloxetina, entre outros. Também diferem nos aspectos farmacocinéticos e no potencial de interação com outras substâncias, tornando-os úteis clinicamente em diversos grupos de pacientes. Para vários antidepressivos novos, a segurança em gestantes e em lactantes ainda não foi estabelecida.
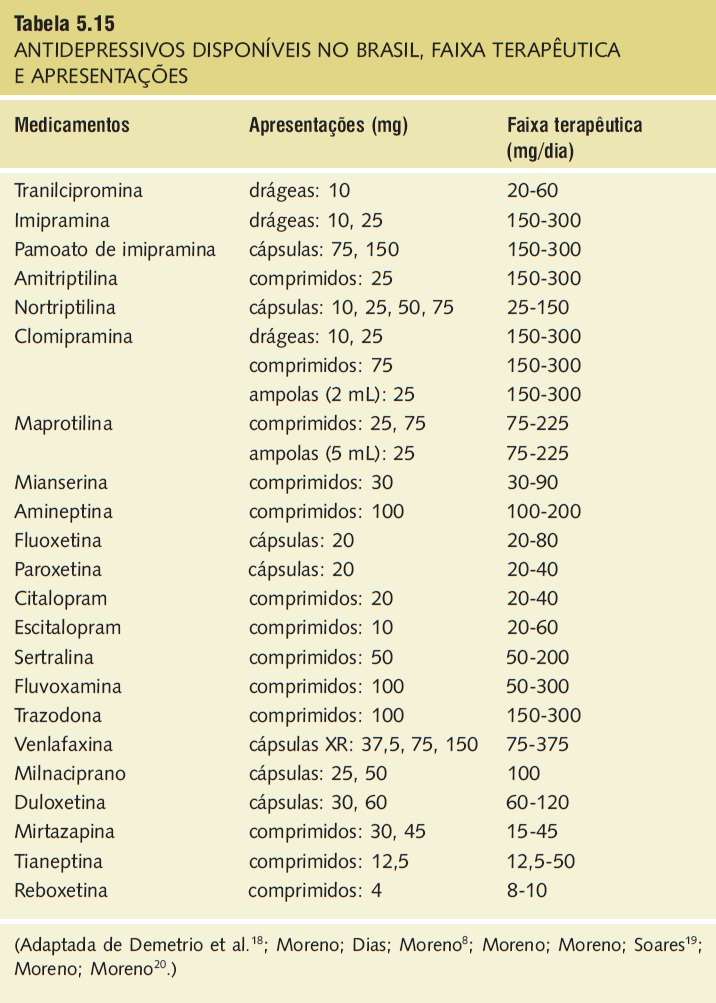
Afirmar que a amplitude do espectro de ação neuroquímica corresponde a um real ganho em termos de eficácia clínica requer testes mais consistentes. As Tabelas 5.13, 5.14 e 5.15 apresentam resumidamente os principais efeitos dos antidepressivos atualmente disponíveis na neurotransmissão, a ação nos diferentes receptores, bem como as apresentações e as faixas terapêuticas preconizadas, respectivamente.
Tabela 5.12
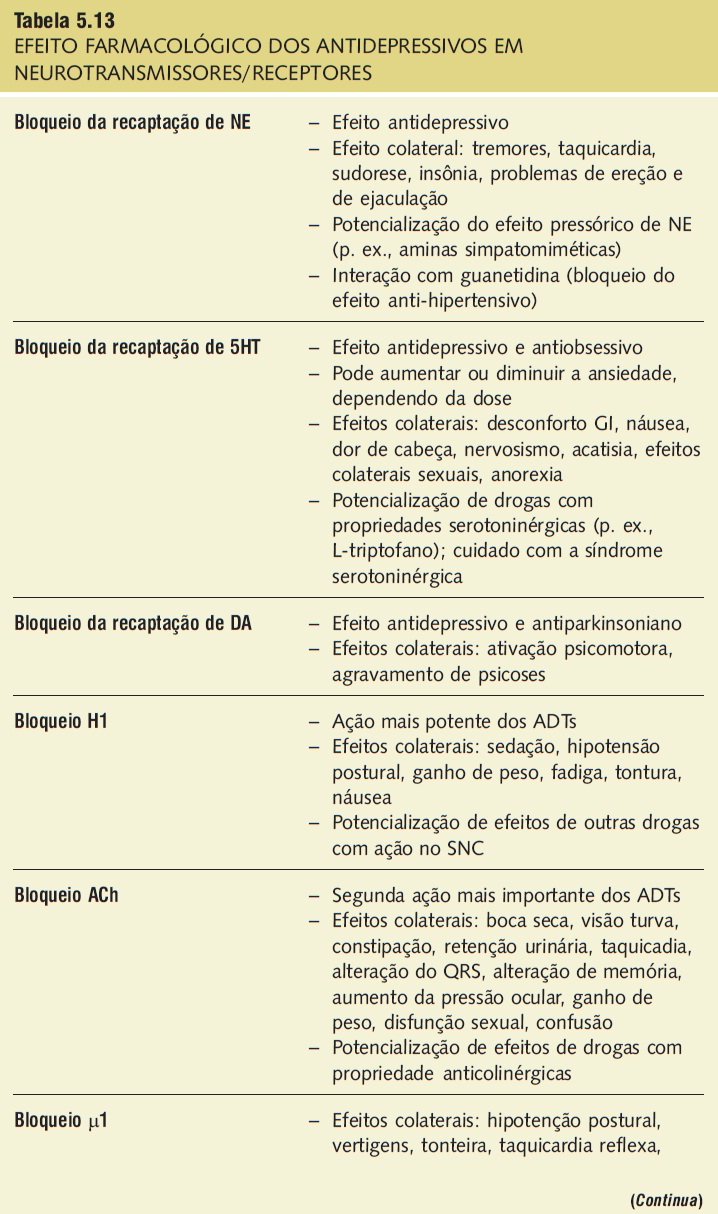
Tabela 5.13
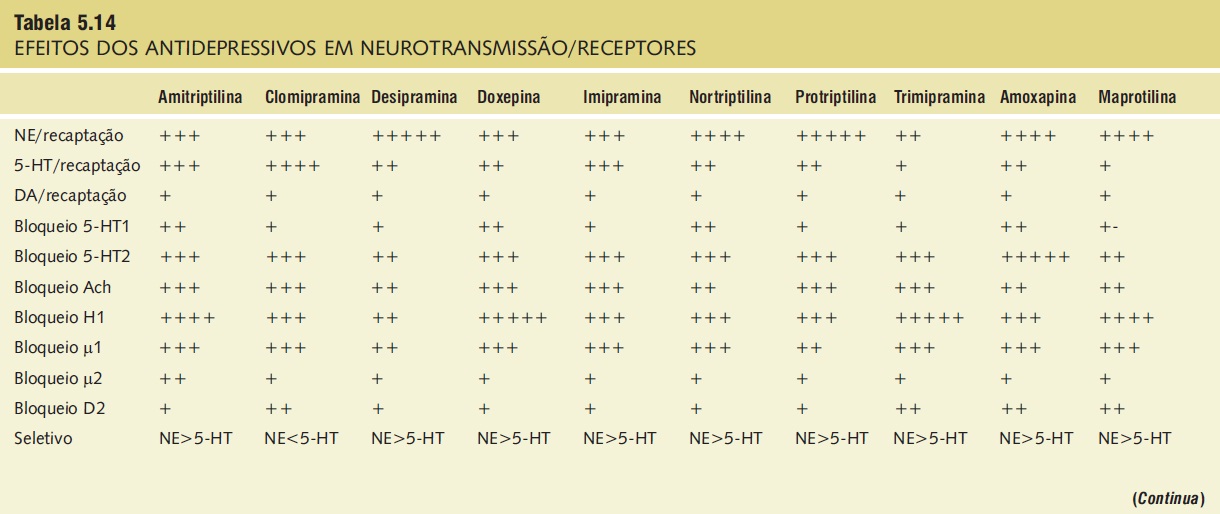
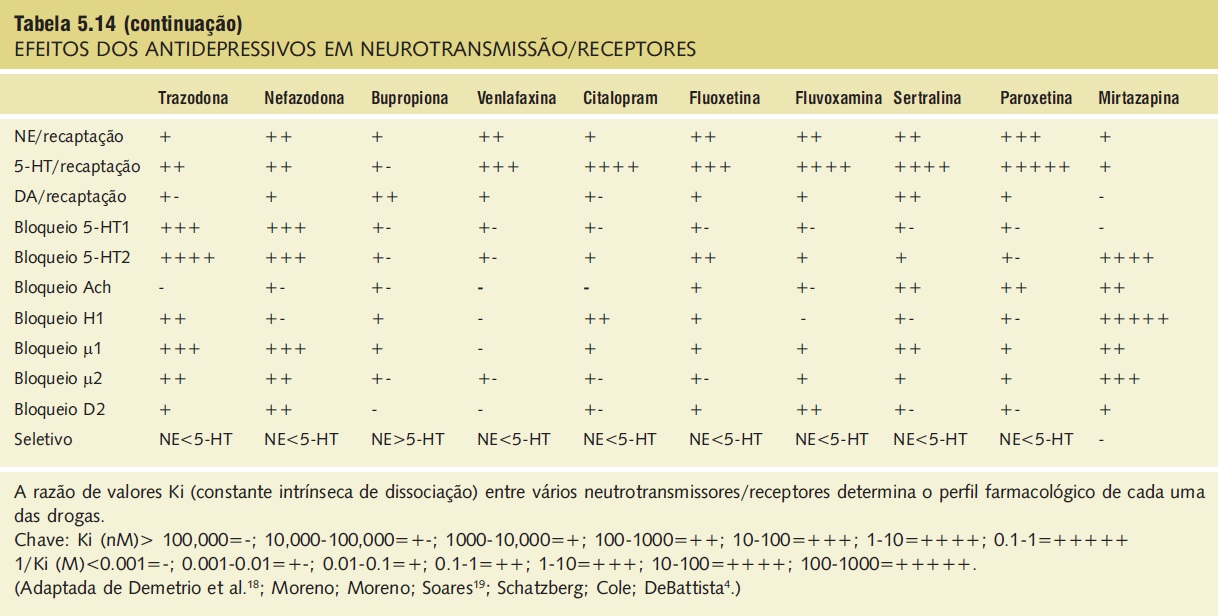
Tabela 5.14
Tabela 5.15
1. O’Reardon JP, Amsterdam JD. Treatment-resistant depression: progress and limitations. Psychiatric Annals. 1998;28:633–40.
2. Kessler RC, Berglund P, Demler O, Jin R, Walters EE. Lifetime prevalence and age-onset distributions of DSM-IV disorders in the national comorbidity survey replication. Arch Gen Psychiatry. 2005 Jun;62(6):593-602.
3. Crown W, Finkelstein S, Brendt E, Ling D, Poret AW, Rush AJ, et al. The impact of treatmentresistant depression in health care utilization and costs. J Clin Psychiatry. 2002;63: 963–71.
4. Schatzberg AF, Cole JO, DeBattista C. Manual of clinical psychopharmacology. 5th ed. Arlington: American Psychiatry Publishing; 2005. p. 239-61.
5. Judd LL. The clinical course of unipolar major depressive disorders. Arch Gen Psychiatry. 1997;54:989–91.
6. Rush AJ, Trivedi MH, Wisniewski SR, Stewart JW, Nierenberg AA, Thase ME, et al. Bupropion-SR, sertraline, or venlafaxine-XR after failure of SSRIs for depression. N Engl J Med. 2006; 354 (12): 1231-42.
7. Kupfer DJ. Long-term treatment of depression. J Clin Psychiatry. 1991;52:28-34.
8. Moreno DH, Dias RS, Moreno RA. Transtornos do humor. In: Neto MRL, Elkis H. Psiquiatria básica 2ª ed. Porto Alegre: Artmed; 2007. p. 219-34.
9. Moreno DH, Moreno RA. Tratamento da depressão resistente a tratamento. In: Oliveira IR, de Sena EP. Manual de psicofarmacologia clínica. 2ª ed. Rio de Janeiro: Guanabara & Koogan; 2006. p. 142-9.
10. Amsterdam JD. SSRI efficacy in severe and melancholic depression. J Psychopharmacol. 1998;12(suppl B):S99-S111.
11. Ayuso-Gutierrez JL. Depressive subtypes and efficacy of antidepressive pharmacotherapy. World J Biol Psychiatry. 2005;6(Suppl 2):31-7.
12. Fawcett J, Barkin RL. Review of the clinical studies on the efficacy, safety and tolerability of mirtazapine for the treatment of patients with major depression. J Affective Disord. 1998;51:267-86.
13. Fleck MP, Moreno RA, Uint L, Andrade AG, Bottino CMC, Kerr-Corrêa F. Efficacy of milnacipram in outpatients experiencing major depression non respondent to SSRIs: a 12-week open study. No prelo 2007.
14. Gentil V, Kerr-Corrêa F, Moreno R, Busnello ED’A, De Campos JA, Juruema MF, et al. Double-blind comparison of venlafaxine and amitryptiline in outpatients with major depression with or without melancholia. J Psychopharmacol. 2000;4(1):61-6.
15. McGrath PJ, Stewart JW, Fava M, Trivedi MH, Wisniewski SR, Nierenberg AA, et al. Tranylcypromine versus venlafaxine plus mirtazapine following three failed antidepressant medication trials for depression: a STAR*D report. Am J Psychiatry. 2006;163(9):1531-41.
16. Trivedi MH, Fava M, Wisniewski SR, Thase ME, Quitkin F, Warden D, et al. Medication augmentation after the failure of SSRIs for depression. N Engl J Med. 2006;354(12):1243-52.
17. Versiani M, Moreno R, Ramakers-van Moorsel CJ, Schutte AJ. Comparison of the effects of mirtazapine and fluxetine in severely depressed patients. CNS Drugs. 2005;19 (2):13746.
18. Demetrio FN, Teng CT, Minatogawa TM, Rocco PTP. antidepressivos. In: Neto MRL, Elkis H. Psiquiatria básica. 2ª ed. Porto Alegre: Artmed; 2007. p. 547-88.
19. Moreno RA, Moreno DH, Soares MBM. Psicofarmacologia de antidepressivos. Rev bras psiquiatr. 1999, 21(2):24-40.
20. Moreno RA, Moreno DH. antidepressivos tricíclicos. In: Cordás TA, Moreno RA, editores. Condutas em psiquiatria. 3ª ed. São Paulo: Lemos; 1999. p.135-61.
21. Fava M. Diagnosis and definition of treatment-resistant depression. Biol Psychiatry. 2003;53(8):649-59.
22. Judd LL, Paulus MJ, Schettler PJ, Akiskal HS, Endicott J, Leon AC, et al. Does incomplete recovery from first lifetime major depressive episode herald a chronic course of illness? Am J Psychiatry. 2000;157(9):1501-4.
23. Ciraulo DA, Shader RI, Greenblatt DJ, Creelman W. Manual de interações medicamentosas em psiquiatria. 3ª ed. Baltimore (Maryland): Lippincott Williams & Wilkins; 2006.
24. Kaplan HI, Sadock BJ. Synopsis of psychiatry. 8th ed. Baltimore: Williams & Wilkins; 1998.
25. Bezchlibnyk-Butler KZ, Jeffries JJ. Clinical handbook of psychotropic drugs. 9th ed. Seattle: Hogrefe & Huber; 1999.
26. Feighner JP. Mechanism of action of antidepressant medications. J Clin Psychiatry. 1999;60( suppl 4):4-11.
27. Himmelhoch JM. Monoamine oxidase inhibitors. In: Kaplan HI, Sadock BJ, editors. Comprehensive textbook of psychiatry. 6th ed. Baltimore: Williams & Wilkins; 1995. p. 2038-56.
28. Goodnick PJ, Goldstein BJ. Selective serotonin reuptake inhibitors in affective disorders – I: Basic pharmacology. J Psychopharmacol. 1998;12(3 suppl B):S3-S20.
29. Malberg JE, Eisch AJ, Nestler EJ, Duman RS. Chronic antidepressants administration increases granule cell neurogenesis in hippocampus of the adult male rat. Soc Nuerosci Abstr. 1999;25:1029.
30. Melo M, Moreno RA. Inibidores da monaminoxidase (IMAOs): qual a melhor dieta? BOTA. 1998;3:4-5.
31. United States Pharmacopeia. Dispensing Information (USP-DI). Drug information for the health care Professional. Massachusetts: World Color Book Services; 1999.
32. Dewan MJ, Anand VS. Evaluating the tolerability of the newer antidepressants. J Nerv Ment Dis. 1999;187:96-101.
33. Healy D, Healy H. The clinical pharmacologic profile of reboxetine: does it involve the putative neurobiological substrates of weelbeing? J Affect Disord 1998;51:313-22.
34. Goldstein BJ, Goodnick PJ. SSRIs in the treatment of affective disorders – III. Tolerability, safety and pharmacoeconomics. J Clin Psychopharmacol. 1998;12(3 suppl B):S55-S88.
35.Horst WD, Preskorn SH. Mechanism of action and clinical characteristics of three atypical antidepressants: venlafaxine, nefazodone, bupropion. J Affective Disord. 1998;51:237-4.
36.Quarantini LC, Lacerda AL, Raskin J, Assunção S, Kerr-Correa F, Mota JI, et al.Long-term safety and tolerability of duloxetine in brazilians with major depressive disorder. No prelo 2007.
37.Montgomery SA. Reboxetine: additional benefits to the depressed patient. J Psychopharmacol. 1997;11(4 suppl):S9-S16.
38.Stahl SM. Psychopharmacology of antidepressants. London: Martin Dunitz; 1997.
39.Bauer M, Bschor T, Pfenning A, Whybrow PC, Angst J, Versiani M, et al. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of unipolar depressive disorders in primary care. World J Biol Psychiatry. 2007;8(2):67-104.
40.Hamon M, Bourgoin S. Pharmacological profile of antidepressants: a likely for their efficacy and side effects? Eur Neuropshycopharmacol. 2006;16:S625-32.
41.Mahli GS, Moore J, McGriffin P. The genetics of major depression disorder. Curr Psychioatry Rep. 2000;2:165-9.
42.Kafitz KW, Rose CR, Thoenen A, Konnerth A. Neutrophin-evoked rapid excitation through TrkB receptors. Nature. 1999 Oct 28;401(6756):918-21.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.